Das “erweiterte” Neugeborenenscreening
Eine erste Untersuchung erfolgt im Normalfall bereits kurz nach der Geburt. Am zweiten oder dritten Lebenstag wird dem Neugeborenen etwas Blut aus der Ferse entnommen und auf die Filterfelder einer Trockenblutkarte (manchmal ist hierbei auch die veraltete Bezeichnung “Guthrie”-Karte zu hören, nach einem früher mal gebräuchlichen Verfahren) getropft. Diese Karte wird mit den Daten des Kindes versehen und an das zuständige Screeningzentrum geschickt. Dort erfolgt die Analyse der im Blut enthaltenen Acylcarnitine mittels eines Verfahrens namens Tandem-Massenspektrometrie. Liegen die aus dem Trockenblut ermittelten Werte ausserhalb des definierten Normbereichs gesunder Probanden, ergibt sich daraus ein erster Verdacht. Zu diesem frühen Zeitpunkt stellt das Testergebnis aber wirklich noch keine verlässliche Diagnose, sondern lediglich einen Verdacht dar, der durch weitere Untersuchungen abgeklärt werden muss.
Das Screening ist ein kostengünstiger Schnelltest, nicht mehr. Es ist vergleichbar mit der Sicherheitskontrolle der Passagiere am Flughafen. Beim Durchschreiten des Metalldetektorbogens leuchtet oben ein rotes Licht auf, wenn der kontrollierte Passagier noch metallene Gegenstände am Körper bei sich trägt. Ob das aber ein in der Hosentasche vergessener harmloser Schlüsselbund ist, oder doch eine Waffe, ist anhand dieses Schnelltests nicht festzustellen, aber die Sicherheitskontrolleure werden durch das rote Licht darauf aufmerksam gemacht, dass nun noch einmal genauer hingeschaut werden muss. Genauso verhält es sich mit dem ersten Ergebnis des Screenings. Eine Diagnose wird aus diesem Anfangsverdacht erst, wenn durch die zueinander passenden Ergebnisse aller weiteren und sehr viel genaueren Untersuchungen das Vorliegen des MCAD-Mangels zweifelsfrei bestätigt wird, oder zumindest mit sehr großer Wahrscheinlichkeit anzunehmen ist. Das ist jedenfalls der offizielle Weg, der dir bzgl. der Diagnosestellung bevorsteht, denn zu einer klaren Aussage wird man sich von ärztlicher Seite aus erst dann durchringen, wenn die insgesamt zur Abklärung definierten Untersuchungsschritte alle durchlaufen wurden. Trotzdem geht bereits aus diesem allerersten Screening in den meisten Fällen schon sehr deutlich hervor, ob dein Kind tatsächlich einen als solchen zu bezeichnenden MCAD-Mangel hat oder nicht!
Der klassische, also schwere MCAD-Mangel ist im Screeningergebnis fast immer direkt zu erkennen, denn bei den dabei üblichen sehr hohen Acylcarnitinwerten besteht keine andere Interpretationsmöglichkeit, als dass dein Kind von beiden Elternteilen eine schwerwiegende MCAD-Mutation vererbt bekommen hat. Auch die sich aus dem häufigsten milden MCAD-Mangel ergebenden Screeningwerte liegen fast immer in einem deutlich zu erkennenden und von dem schweren MCAD-Mangel zu unterscheidenden niedrigeren Bereich.
Die von allen Familien erhoffte Möglichkeit, dass es sich bereits im Kontrollscreening aufgrund dann bereits normalisierter Werte als falscher Alarm (aufgrund der durch einen reinen Carrierstatus anfangs nur sehr leicht erhöhten Werte) herausstellen kann, ist aufgrund bestimmter Wertekonstellationen ebenfalls mit sehr hoher Wahrscheinlichkeit bereits dem ersten Screeningergebnis zu entnehmen. In seltenen Fällen kann es sich trotz tatsächlich dann bereits im Recall normalisierter Werte – was für viele Kinder mit MCAD-Anfangsverdacht daraufhin zur vollständigen Entwarnung führt – durch die an manchen Kliniken auch noch direkt durchgeführten weiteren Abklärungsschritte (genetische und enzymatische Untersuchung) ergeben, dass doch eine zweite Mutation im MCAD-Gen gefunden wird, die aufgrund einer hohen Restaktivität, die sich bereits durch die nur gering erhöhten Werte des Screenings gezeigt hat, vermutlich als harmlos angesehen werden kann. Dennoch würde in diesem Fall – aufgrund der nun mal gefundenen zwei Mutationen – die Diagnose (milder) MCAD-Mangel erfolgen und auf Dauer bestehen bleiben.
Kurz gesagt, lässt sich aus den Werten des ersten Screenings in den allermeisten Fällen bereits sehr deutlich heraus die Unterscheidung zwischen
- definitiv ein schwerer MCAD-Mangel
- wahrscheinlich kein schwerer, aber vermutlich doch ein milder MCAD-Mangel
- wahrscheinlich gibt es schon im Recall Entwarnung, aber es könnte sich auch um einen ganz extrem milde Form handeln, falls doch eine zweite Mutation gefunden werden sollte, die dann halt trotzdem MCAD-Mangel genannt wird.
Acylcarnitine
Diese ganzen hinsichtlich des Ausschlusses oder Auffindens eines MCAD-Mangels im Screening auf ihre Konzentrationen hin überprüften Acylcarnitine C6, C8, C10 und C12, sowie verschiedene Quotienten dieser Werte, sind − vereinfacht ausgedrückt − die an Carnitin gekoppelten, aus den Zellen abtransportierten mittelkettigen Fettsäurenreste mit 6er, 8er, 10er oder 12er Länge. Diese konnten aufgrund des MCAD-Mangels nicht verarbeitet werden und müssen nun aufgrund ihrer ansonsten toxischen Wirkung auf verschiedene Organe den Körper auf dem Weg über die Nieren unbedingt wieder verlassen. Überschreitet die Konzentration dieser Substanzen im Blut den zugrunde gelegten Normbereich, deutet dies darauf hin, dass die Verarbeitung der mittelkettigen Fettsäuren innerhalb der Zellen nicht vollständig funktioniert, weshalb diese Reste nun in größerer Menge über das Blut abtransportiert werden. Dies begründet dann den Verdacht auf einen vorliegenden MCAD-Mangel. Richtungsweisend ist hier primär der deutlich erhöhte C8-Wert.
Ein für den MCAD-Mangel typisches Acylcarnitinprofil sieht z.B. so aus:
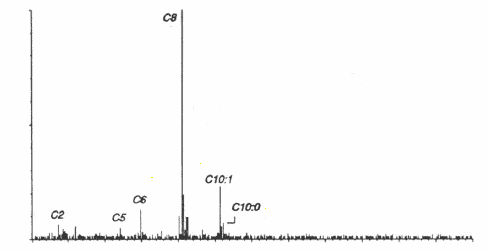
| zur Erläuterung: die Zahlenwerte der Größenachse wurden von mir absichtlich entfernt, da die Konzentration von C8 bei jedem MCAD-Patienten völlig unterschiedlich ausfallen kann. Auffällig ist aber grundsätzlich der gegenüber den umgebenden C6 und C10 deutlich hervorstechende Ausschlag der Messwerte beim C8 (Octanoylcarnitin). C6 und C10 sind üblicherweise auch nachweisbar erhöht und unterhalb von C6 finden sich nur noch sehr geringe Konzentrationen. Ein “normales” Acylcarnitinprofil hat dagegen sehr niedrige Werte bei C6, C8 und C10, während gleichzeitig ein sehr hoher Ausschlag bei C2 und ein leichter bei C4 existiert. Dies zeigt, dass die ursprünglich langkettigen Fettsäuren bereits nahezu vollständig in die 2er Kohlenstoffketten (Ketonkörper) aufgespalten wurden. |
Das Screeningzentrum übermittelt den Befund der Trockenblutkarte zurück an den Einsender. Das ist die Person oder Einrichtung, die für die Betropfung der Karte verantwortlich war. Dabei kann es sich um die Säuglingsstation des Krankenhauses, aber auch um den Kinderarzt oder um die von den Eltern alleine für die Geburtsbegleitung beauftragte Hebamme handeln. Diese müssen rund um die Uhr erreichbar sein, da sie danach für die sofortige Mitteilung des auffälligen Befundes an die Eltern des betreffenden Kindes verantwortlich sind. Üblicherweise werden Eltern nur dann informiert, wenn das Screening ein auffälliges und somit kontrollbedürftiges Ergebnis erbracht hat. Unauffällige Befunde gehen zwar auch zurück an den Einsender, werden den Eltern aber nur auf gezielte Nachfrage hin mitgeteilt. Die Eltern werden nach einer auffälligen Rückmeldung schnellstmöglich über den vorliegenden Verdacht, die weiteren Schritte und die auf jeden Fall schon mal vorsorglich einzuhaltenden Maßnahmen informiert werden. Mehr als ein “Die Werte des Neugeborenen können auf einen MCAD-Mangel hindeuten, bitte informieren Sie die Eltern, und sagen Sie ihnen, dass sie das Kind regelmäßig füttern müssen, ansonsten kann es ins Koma fallen oder sogar sterben!” bleibt aber selten bei den zuständigen und bzgl. des MCAD-Mangels meist völlig ahnungslosen Mitarbeitenden der Station hängen. Daher ist die von ihnen direkt danach weitergegebene Kurzinformation an die Eltern zwar wichtig (auch wenn bei dieser Gelegenheit so gut wie alle bei den geschockten Eltern aufkommenden Fragen unbeantwortet bleiben und dadurch immer eine Menge Ängste hervorgerufen werden), darf aber noch nicht als endgültige Schicksalsentscheidung betrachtet werden.
Haben sich Auffälligkeiten im Screening gezeigt, wird der Befund auch sofort an den von den Eltern benannten Kinderarzt weitergeleitet, mit dem man sich dann möglichst schnell zwecks Kontrolluntersuchung in Verbindung setzen sollte. Keine Panik! Solange das Kind gesund ist, und schon annähernd regelmäßig trinkt, macht es nichts, wenn sich dieser Termin noch um ein paar Tage verzögert. Nur die Nerven der zu dem Zeitpunkt üblicherweise noch sehr wenig oder sogar völlig uninformierten und daher stark besorgten Eltern werden bis dahin möglicherweise schon auf eine harte Geduldsprobe gestellt.
In die Erstinformation und -beratung sollte man auch nicht mit zu großen Erwartungen gehen! Mit immer noch sehr großer Wahrscheinlichkeit hat auch der betreffende Kinderarzt noch nie zuvor von dem MCAD-Mangel gehört, geschweige denn einen davon betroffenen Säugling in seiner Praxis gehabt und sich sein “Fachwissen” noch schnell über Nacht angelesen − immerhin. Der Kinderarzt ist für fast alle medizinischen Belange der erste und richtige Ansprechpartner, aber in Bezug auf eine so spezielle und seltene Stoffwechselstörung sollte man von ihm keine umfassenden Kenntnisse erwarten. Im besten Fall ist er beim Googeln auf mcad-infos.de gestoßen und hat hier tatsächlich ein bis zwei Stunden in das schnelle Einlesen in die Thematik investiert. Wahrscheinlicher ist aber, dass er die meist nur sehr knappen und für die Eltern kaum hilfreichen Informationen auf den Seiten einiger deutscher Uni-Klinken überflogen und kurz auf den Zettel mit den Empfehlungen für die weiteren Schritte geschaut hat, die er zusammen mit dem Befund vom Screeningzentrum bekommen hat. Diese besagen z.B., dass solch ein durch ein auffälliges Erstscreening aufgekommener Verdacht grundsätzlich noch durch ein zweites Kontrollscreening − den sogenannten Recall − bestätigt (“konfirmiert”) werden muss. Zeigt auch dieses auffällige Werte, ist eine zügige weitere Abklärung im Stoffwechselzentrum angeraten. Eine endgültige Sicherung der Diagnose kann z.B. durch Messung der Enzymfunktion oder durch eine molekulargenetische Analyse erfolgen. Beim MCAD-Mangel wurde bis vor einigen Jahren hauptsächlich die Genanalyse durchgeführt, da man inzwischen eine Anzahl bestimmter Mutationen des ACADM-Gens für diese Stoffwechselstörung verantwortlich machen konnte. Inzwischen wird dagegen meist die Messung der Residualaktivität des MCAD-Enzyms als erster weiterer Diagnoseschritt durchgeführt, weil man dadurch auch beim Vorliegen von seltenen und somit oft noch hinsichtlich ihrer Auswirkung unbekannten Genmutationen direkt herausfinden kann, wieviel das MCAD-Enzym im Vergleich zu seiner normalen Form noch zu leisten in der Lage ist. Manchmal wird dann als weiterer Schritt zur Absicherung der Diagnose auch noch die genetische Mutationsbestimmung empfohlen. Allerdings bringt sie dann keinen echten Mehrwert mehr, denn wenn die Residualaktivität bereits bei 0-5% ermittelt wurde, spielt es auch keine Rolle mehr, wenn bei dem Kind dann vielleicht doch nicht die häufigste und als schwere MCAD-Ausprägung bekannte Variante K329E homozygot gefunden würde. Egal welche Mutationskombination letztlich bei ihm vorliegt, ihr Schweregrad wäre dem der zuvor genannten Hochrisikovariante absolut vergleichbar. Besonders dann, wenn die Krankenkasse eine solche zusätzliche genetische Untersuchung nicht mehr übernehmen will, kann man getrost darauf verzichten und sollte diese rund 2000€ kostende Untersuchung auch nicht aus eigener Tasche bezahlen. Etwas anderes ist es natürlich, wenn die Uni-Klinik diese Untersuchung kostenlos anbietet, weil z.B. gerade jemand in diesem Bereich forscht und man dringend Daten generieren muss.
Bitte nicht nachmachen!
Zur Durchführung einer Genanalyse bei einem Kind, z.B. zwecks Abklärung einer Stoffwechselstörung, ist streng genommen eine handschriftlich unterschriebene Einverständniserklärung beider Elternteile erforderlich, obwohl ich schon von einigen Eltern erfahren habe, dass sie anscheinend einfach nur mitgeteilt bekommen hatten, dass eine Genanalyse gemacht würde, ohne dass sie diesbezüglich um ihre Zustimmung, geschweige denn um ihre Unterschrift gebeten worden seien. Wie auch immer – leider gibt es doch immer wieder vereinzelt Eltern, die diese Genanalyse oder die Enzymanalyse verweigern. Vielleicht fürchten sie sich vor der letzten Bestätigung der Diagnose und ziehen es deshalb vor, im Ungewissen zu bleiben: Man weiß es ja nicht wirklich, also hat das Kind auch nichts! “Alle meine Freunde und auch mein (irgendwas)-Arzt sagten mir doch immer, dass mein Kind vollkommen gesund aussieht!” ist vermutlich einer der häufigsten Sätze, den die Ärzte der Stoffwechselzentren dann irgendwann zu hören bekommen. Das ist das große Problem! Man sieht Kindern den MCAD-Mangel nicht an, sondern kann ihn nur durch aufwändige Untersuchungen nachweisen.
Messung der Enzymaktivität
Zur alternativen Kontrolle der Enzymfunktion gibt es heutzutage sehr zuverlässige Möglichkeiten, die sogenannte Residualaktivität des MCAD-Enzyms aus einer kleinen Blutprobe zu messen. Wie das inzwischen an mehreren deutschen Uni-Kliniken, vor allem in Freiburg, durchgeführte Verfahren im Detail funktioniert, ist mir nicht bekannt, daher kann ich an dieser Stelle dazu auch keine Informationen weitergeben.
Früher, also Anfang der 2000er, wurde die verbleibende Restaktivität des MCAD-Enzyms nicht aus einer einzuschickenden Blutprobe, sondern in einem mehrstündigen Test direkt am Kind ermittelt, z.B. durch den Hexanoyl-CoA-Substrat- oder Phenylpropionsäuretest. Ähnlich dem Zuckerbelastungstest, den manche Frau während der Schwangerschaft aufgrund des Verdachts auf einen Gestationsdiabetes machen lässt, wurde damit getestet, ob vom kindlichen Organismus MCAD-Enzyme gebildet wurden, bzw. wie groß deren Aktivität im Vergleich zu den Normwerten bei Kindern ohne MCAD-Mangel war. Im Normalfall wurden dann nämlich die von dem Kind zu schluckenden 25-50mg/kg Hexanoyl-CoA-Substrat oder Phenylpropionsäure durch das MCAD-Enzym aufgespalten. Bestand tatsächlich ein MCAD-Mangel, wurde die Phenylpropionsäure an Glycin gekoppelt und im Urin als nachweisbares Phenylpropionglycin ausgeschieden. Der Test wurde für gewöhnlich morgens durchgeführt. Vor der Verabreichung der berechneten Dosis Phenylpropionsäure wurde zunächst eine Vorher-Urinprobe genommen. Da die Testsubstanz einen unangenehmen Zimtgeschmack hatte, konnte sie in Tee eingerührt, oder z.B. auch mit etwas Marmelade vermischt werden. Danach durfte das Kind ganz normal essen und trinken. Der Urin wurde über einen Zeitraum von 6 bis 12 Stunden nach der Einnahme gesammelt und auf das Vorhandensein der entsprechenden organischen Säuren hin analysiert. Dieser alte Test wurde fast vollständig durch die Tandem-Massenspektrometrie abgelöst, da mit dieser äusserst sensibel eingestellten Analysemethode inzwischen schon mehrere “milde” MCAD-Varianten im Nachhinein diagnostiziert wurden, die bei einem zuvor durchgeführten Phenylpropionsäuretest unentdeckt geblieben waren.
Diese etwas veraltet erscheinende Information ist für dich unter Umständen aber trotzdem wichtig zu wissen, da mir auch heute noch immer mal wieder Eltern davon berichten, dass ihr Stoffwechselarzt eine Residualaktivitätsanalyse rigiros ablehne, mit der Begründung, das Verfahren sei seit langem überholt und tauge auch nichts. Vermutlich ist er mit seinem diesbezüglichen Kenntnisstand noch bei dem gerade beschriebenen alten, wirklich sehr ungenauen und daher schon lange nicht mehr durchgeführten Verfahren stehengeblieben und kennt nicht die völlig anders funktionierenden und sehr viel besseren aktuellen Methoden zur Einschätzung der Schwere eines MCAD-Mangels..
Die oben schon erwähnten präziseren Testmethoden für die Feststellung der Restaktivität des MCAD-Enzyms werden zur Zeit in Deutschland z.B. im Klinikum der Unis Freiburg, Düsseldorf und Heidelberg an den dort betreuten MCAD-Patienten durchgeführt und ständig weiterentwickelt. Anhand der gemessenen Enzymaktivitäten lässt sich meist relativ sicher zwischen milden und schweren Ausprägungen des MCAD-Mangels unterscheiden. Nach wie vor macht aber das Ergebnis für die meisten Stoffwechselärzte keinen Unterschied bzgl. der den Eltern empfohlenen Behandlungsweise des Kindes. Zumindest aber bietet die Residualaktivitäts-Messung bei deutlich höheren Messwerten ein weiteres starkes Indiz, um die wenigen im Recall noch nicht ausgeschlossenen falsch-positive MCAD-Befunde nachträglich identifizieren zu können.
Manchmal kann auch die Kombination von Tandem-Massenspektrometrie (TMS) und anschließender Mutationsanalyse (s.u.) keine eindeutige Diagnose liefern, z.B. dann, wenn das Neugeborenenscreening ein hinsichtlich des MCAD-Mangels auffälliges Acylcarnitinprofil zeigt, bei der anschließenden Gensequenzierung aber nur eine heterozygote, also nur von einem einzigen Elternteil beigetragene Mutation gefunden werden kann und weitere Screenings ein sich zunehmend normalisierendes Ergebnis zeigen. In diesen widersprüchlichen Fällen sollte man als Eltern, auch wenn dies von dem Arzt zuvor nicht angedacht wurde, auf die zusätzliche Durchführung eines Enzymfunktionstests (s.o.) bestehen, denn nur damit kann dann wirklich festgestellt werden, ob MCAD-Enzyme gebildet werden, und was diese zu leisten in der Lage sind. Manche deutsche Ärzte halten diese weitere Abklärung zwar für überflüssig, für die betroffenen Eltern kann es aber sehr wichtig und vor allem beruhigend sein, endlich Klarheit über den Befund ihres Kindes zu bekommen − egal wie das Ergebnis am Ende ausfällt. In den Niederlanden ist die Messung der Enzymaktivität nach wie vor ein wichtiger Standard-Diagnoseschritt und wird üblicherweise gleich nach einem auffälligen Screeningbefund durchgeführt, noch bevor man nach irgendwelchen Mutationen des ACADM-Gens sucht.
Ganz allgemein führen auch rein heterozygote Mutationen bei rezessiv vererbbaren Enzymdefekten bereits zu einer deutlichen Aktivitätsminderung des betreffenden Enzyms. Im Vergleich zu der bei nicht betroffenen Personen messbaren normalen Aktivität liegt sie bei heterozygoten Carriern nur bei etwa 50-80%, je nach Schwere des jeweiligen Gendefekts. Diese verminderte Enzymleistung reicht aber vollkommen aus, so dass diese Menschen niemals Gefahr laufen, selbst eine Stoffwechselentgleisung zu erleiden. Das kommt daher, dass im Normalfall die meisten Enzyme im menschlichen Körper in einer Konzentration weit über dem tatsächlich benötigten Maß zur Sicherstellung eines ausgeglichenen Stoffwechsels vorliegen. Dadurch führt auch eine Aktivitätsminderung um 50% noch zu einem weitgehend völlig normalen metabolischen Durchlauf, der sich kaum von dem eines Menschen mit zwei Wildtyp-Allelen unterscheidet. Die dennoch bestehenden kleinen Unterschiede lassen sich nur mit aufwändigen Untersuchungsmethoden herausfinden, spielen im Leben des Carriers aber keine Rolle. Milde MCAD-Varianten, denen inzwischen kaum noch ein Krankheitswert zugesprochen wird (z.B. Y67H mit K329E compound heterozygot) ergeben bei dem in Deutschland durchgeführten Analyseverfahren eine Residualaktivität von um die 20%, was noch einmal zeigt, dass die “normalen” 100% des Menschen mit zwei Wildtyp-Allelen nicht als die gerade so ausreichende Menge, sondern als weit überdimensioniert angesehen werden können.
Viele falsch-positive Befunde beim Neugeborenenscreening
Aufgrund der hohen Sensitivität der TMS führt das Neugeborenenscreening jedoch auch sehr häufig zu einem vermeintlich auffälligen MCAD-Befund, der sich oft schon durch ein nach einigen Tagen erfolgendes Kontrollscreening (Recall) mit dann unauffälligem Ergebnis als unbegründet herausstellt. Meist handelte es sich dabei um Verdachtsfälle, bei denen die im Erstscreening ermittelten Acylcarnitinwerte auch schon nur geringfügig über den zugrunde gelegten Normbereichen lagen.
Die Proben für das Neugeborenenscreening werden zu einem relativ frühen Zeitpunkt, etwa 36-72 Stunden nach der Geburt, abgenommen. Dies ist auch notwendig, da in dem Screening gleichzeitig noch eine ganze Reihe weiterer Stoffwechselstörungen getestet werden, für die später entnommene Blutproben keine ausreichende Diagnosegenauigkeit mehr erlauben würden. Da sich zu diesem Zeitpunkt ein beträchtlicher Anteil der Neugeborenen noch in einer deutlichen katabolen Stoffwechselsituation befindet, ist besonders die bei heterozygoten Carriern zu erwartende niedrigere MCAD-Enzymaktivität oft nicht ausreichend, um eine vollständig normale Fettsäuren-Homöostase aufrecht zu erhalten. Dadurch werden auch solche Kinder manchmal im ersten Screening und vereinzelt auch noch im Kontrollscreening aufgrund leicht erhöhter Werte auffällig, obwohl bei ihnen kein MCAD-Mangel vorliegt, die Fettsäuren-Verarbeitung schon nach wenigen Tagen völlig normal ablaufen wird und vermutlich zu keinem Zeitpunkt die Gefahr einer Stoffwechselentgleisung besteht.
Da man aber zu diesem frühen Zeitpunkt eine zwar milde, aber trotzdem behandlungsbedürftige Variante des MCAD-Mangels als Ursache für leicht erhöhte Screeningwerte nicht sicher ausschließen kann, müssen alle diese Fälle in einem Zweitscreening, und bei weiterhin bestehendem Verdacht auch mittels einer molekulargenetischen Untersuchung bzw. Residualaktivitäts-Messung abgeklärt werden.
Wie eine niederländische Studie über zwei Jahre zeigte, kann der beim Screening untersuchte C8:0-Wert (Octanoylcarnitin-Konzentration) zwischen dem ersten und dem zweiten Screening auch bei Vorliegen eines echten MCAD-Mangels beträchtlich absinken, jedoch bleibt z.B. der ebenfalls bestimmte Quotient C8:0/C10:0 (Verhältnis von Octanoylcarnitin zu Decanoylcarnitin) dann trotzdem auf einem deutlich erhöhten Wert. Damit lassen sich in den meisten Fällen die falsch-positiven Befunde (kein MCAD-Mangel) von den wahr-positiven Befunden (MCAD-Mangel) unterscheiden. Letzte Sicherheit kann aber oft nur die molekulargenetische Mutationsbestimmung bringen.
Ein kleiner Einschub: meine eigene Erfahrung
Im Geburtsvorbereitungskurs warnte uns unsere Hebamme davor, dass die Säuglingsschwestern im Krankenhaus vermutlich sofort aufs Zufüttern mit der Flasche drängen würden, um ein schreiendes Kind nachts zur Ruhe zu bringen. Aufgrund der völlig anderen Saugeigenschaften eines Flaschenschnullers könne dies zu einem so frühen Zeitpunkt aber zu einer nur noch sehr schwer korrigierbaren Saugverwirrung führen, in deren Folge das eigentlich angestrebte vollständige Stillen des Kindes extrem erschwert würde. Statt dessen empfahl die Hebamme das Kind einfach öfter anzulegen. Diesem Rat folgend haben wir das von den Schwestern tatsächlich vehement geforderte Zufüttern konsequent abgelehnt. Unser Sohn wurde also von Anfang an nur gestillt und hat in den ersten drei Tagen, dem noch geringen Nahrungsangebot entsprechend, deutlich an Gewicht verloren.
Im Rückblick ist damit das knapp über dem Normbereich liegende Acylcarnitinprofil im NG-Screening erklärbar. Das Zusammenwirken des noch nicht eingespielten Stoffwechsels mit der stark katabolen Stoffwechsellage und der reduzierten MCAD-Enzymaktivität (aufgrund des inzwischen nachgewiesenen Carrier-Status) musste sich fast zwangsläufig in irgendeiner Weise äussern. Hätten wir dem Drängen der Schwestern nachgegeben und von Beginn an zugefüttert, wäre das Screeningergebnis in unserem Fall wahrscheinlich völlig unauffällig ausgefallen und wir hätten nicht aus unserer eigenen Initiative heraus eine 14-monatige Ärzteodyssee bis zur endgültigen Widerlegung der Erstdiagnose absolvieren müssen. Trotzdem sind wir nach wie vor sicher, alles richtig gemacht zu haben − immerhin entstand aus diesem Anlass auch diese Webseite!
In den 10 Jahren des Forums hatte ich bei mehreren der mir von Teilnehmern zugesandten Screeningbefunden aufgrund der enthaltenen nur gering erhöhten Werte den starken Verdacht, dass es sich dabei ebenfalls nur um einen reinen Carrier-Status des Kindes handeln dürfte, obwohl all diesen Eltern bereits das Vorliegen eines schweren MCAD-Mangels mitgeteilt bzw. in Aussicht gestellt worden war. Auf meine gezielte Nachfrage stellte sich heraus, dass es sich in fast allen Fällen ebenfalls um das erste Kind, folglich um einen verzögerten Milcheinschuss, eine deutliche Gewichtsabnahme aufgrund des nicht erfolgten Zufütterns und eine relativ frühe Blutabnahme für das Screening handelte. Bei einigen dieser Kinder, deren Konfirmationsscreening oder genetische Untersuchung zu dem Zeitpunkt sogar noch ausstand, wurde dann bereits durch das Ergebnis dieser Untersuchungen der Carrier-Status bestätigt. Bei den anderen mussten die Eltern mit etwas Nachdruck die weiteren Untersuchungen zur Abklärung einfordern und der MCAD-Verdacht wurde auf diesem Weg widerlegt. Für drei der Kinder, deren für mich doch zweifelhafte MCAD-Diagnose bereits etwas länger zurück lag, ergab es sich dann bei unterschiedlichen Gelegenheiten (teilweise direkt in der Stoffwechselambulanz beim Kontrolltermin), dass die Eltern in den von ihnen bei ihrem Arzt eingeforderten Befundkopien schließlich den Vermerk fanden, dass der Verdacht auf den MCAD-Mangel bei ihrem Kind eindeutig wiederlegt sei. Ebenso wie bei uns war auch ihnen gegenüber dies zu keinem Zeitpunkt erwähnt worden, sondern sie hatten sogar bei zuvor erfolgten Nachfragen, ob der MCAD-Mangel ihres Kindes denn absolut unumstößlich sei, dies mit aller Deutlichkeit bejaht bekommen. Ein Arzt fuhr die Eltern sogar verärgert an, als sie ihn diesbezüglich zur Rede stellten, dass sie das überhaupt nichts anginge, sie ja keine Stoffwechselexperten seien und sie diese Befundkopien eigentlich überhaupt nicht hätten bekommen sollen. Die restlichen 2-3 Familien mussten schließlich erst an eine andere Uni-Klinik wechseln, um mit einem neuen Arzt die Widerlegung der bestehenden MCAD-Diagnose zu erreichen, da ihr erster Arzt zu keinen weiteren Untersuchungen und zu keinem Überdenken seiner vom ersten Tag an gefassten Meinung bereit war. Diese unrühmlichen Ausnahmen sind aber genau das: unrühmliche Ausnahmen! Pro Jahr gab es unter den neuen Forumsteilnehmern im Durchschnitt nur ein bis zwei Familien, deren Kind fälschlicherweise ein eindeutig bestätigter MCAD-Mangel angedichtet worden war, obwohl bereits der NG-Screeningbefund mit nur leicht erhöhten Werten und unter Berücksichtigung der besonderen Umstände der ersten paar Lebenstage des Kindes sehr stark einen reinen Carrier-Status vermuten lies. Es kommt also wirklich nur vereinzelt vor, dass sich ein vermeintlich als schwer bestätigter MCAD-Mangel (bzw. der den Eltern bereits als eindeutig bestätig mitgeteilt wurde) im Nachhinein doch noch als Irrtum und als reiner Carrier-Status herausstellt. Von alleine passiert es ohnehin nicht, dass der betreuende Stoffwechselarzt seine bisherigen (nicht zutreffenden) Aussagen revidiert, sondern es erfordert eine Menge Eigeninitiative der Eltern mit oftmals sogar dem Wechsel an eine andere Uni-Klinik, um mit Einholung einer zweiten Meinung die ganze Geschichte nochmal neu aufzurollen.
Für den weitaus größten Teil der nur aufgrund ihres möglichen Carrier-Status zunächst leicht auffälligen Neugeborenen wird das vermutete Vorliegen eines MCAD-Mangel schon durch das Kontrollscreening ganz schnell wieder ausgeschlossen. Daher haben es auch damals schon fast alle diese Familien, für die sich das Thema schnell erledigt hatte, ohnehin nicht bis ins Forum geschafft (Ich weiß dies auch nur, da sich über die Jahre hinweg viele dieser Familien direkt nach dem Anfangsverdacht per Email mit ihren Fragen an mich gewandt und sich nicht im Forum registriert hatten), was zeigt, dass die nach Durchführung aller Untersuchungen stehende Diagnose eines MCAD-Mangels normalerweise als sehr verlässlich angesehen werden kann. Trotzdem ist es für die vereinzelt auftretenden, in Wirklichkeit dann doch nicht davon betroffenen Familien um jeden dieser wichtigen frühen Tage, die kommenden Wochen, Monate und Jahre schade, die sie in Angst und Sorge um ihr Kind verbringen, obwohl es keinen Grund dazu gibt.
Besonders heterozygote Carrier ohne MCAD-Mangel fallen oft durch leicht erhöhte Screeningwerte auf!
In Hochrechnungen zufolge rund 70% der falsch-positiven Befunde handelt es sich um die zuvor schon beschriebenen Kinder mit zwar nachweisbarer, aber rein heterozygoter ACADM-Mutation. Auch dabei liefert das Neugeborenenscreening meist zunächst geringfügig erhöhte Acylcarnitin-Werte, jedoch sind diese Kinder lediglich Carrier, also Träger des Defekts − wie etwa jeder 60. Deutsche und auch wenigstens eines seiner Elternteile − aber haben selbst keinen MCAD-Mangel. Der rein heterozygote Carrierstatus ist ohne jegliche klinische Relevanz.
Es scheint jedoch ein selbst bei vielen Stoffwechselärzten verbreiteter Irrtum zu sein, dass bei rein heterozygotem Vorliegen eines MCAD-Gendefekts auch zwingend alle Screeningergebnisse unauffällig sein müssten. Der Gedankengang “Carrier haben keinen MCAD-Mangel, daher werden sie im Screening auch nicht gefunden! Wenn über dem Normbereich liegende Werte festgestellt werden, kann es sich daher nicht um einen Carrier mit heterozygotem und damit harmlosem Gendefekt handeln!” ist daher nicht zutreffend. Carrier können im Screening unauffällig sein, genausogut können sie aber auch mit leicht erhöhten Werten in die Gruppe der auffälligen Befunde geraten. Meistens haben sich die Werte im wenige Tage später folgenden Kontrollscreening bereits normalisiert, manchmal aber auch nicht.
Auf diese in grenzwertigen Fällen noch häufig bestehende Unsicherheit hinsichtlich der Deutung der mittels der TMS gefundenen Analyseergebnisse, wird in anderen Artikeln noch genauer eingegangen. An dieser Stelle soll aber festgehalten werden, dass sich ein großer Teil der (leicht) auffälligen TMS-Erstscreenings nach weiteren Untersuchungen als falscher Alarm herausstellt.
Im Abschlussbericht zum Modellprojekt zur Neuordnung des Neugeborenenscreenings in Bayern wurde festgestellt, dass sich von den ursprünglich 107 auffälligen und kontrollbedürftigen MCAD-Befunden des Erstscreenings ganze 73 als falsch-positiv herausstellten. Bei 34 Kindern bestätigte sich der ursprüngliche Verdacht anhand weiterer Untersuchungen. Der sich aus dem Screening ergebende positive Vorhersagewert bzgl. des tatsächlichen Vorliegens eines MCAD-Mangels lag dieser Studie (Mitte der 2000er) zufolge bei ca 32%. Im Gegenzug bestand daher eine große Chance, dass sich der beim NG-Screening aufgekommene MCAD-Verdacht noch als unbegründet herausstellt. Diese Feststellung liegt jetzt aber schon einige Jahre zurück. In den letzten paar Jahren hat sich laut den jährlichen Screening-Berichten der positive Vorhersagewert mehr in Richtung 50% verschoben. Obwohl jährlich deutlich mehr Kinder geboren werden als z.B. noch 2014, ist die Anzahl der kontrollbedürftigen Erstscreenings in etwa gleich geblieben oder sogar noch gesunken. Von diesen Recalls bestätigt sich der MCAD-Mangel anhand Gensequenzierung oder Enzymanalyse inzwischen bei etwa 50% der Fälle. Es gibt jedoch nicht mehr MCAD-Fälle als noch die Jahre zuvor, sondern die Anzahl der falsch-positiven Fälle wurde durch geeignetere Anpassung der Schwellwerte reduziert. Trotzdem bleibt festzuhalten, dass es immer noch für rund die Hälfte aller kurz nach der Geburt an die Eltern mitgeteilten MCAD-Verdachtsfälle im Verlauf der Konfirmationsdiagnostik Entwarnung geben wird.
In den meisten dieser sich als falsch-positiv herausstellenden Fällen dürfte es sich um Carrier-Kinder handeln. Dies kann jedoch nur vermutet werden, da es bei diesen Kindern − mit sehr wenigen Ausnahmen − gar nicht erst zu einer molekulargenetischen Untersuchung kommt, in der der rein heterozygot vorliegende Gendefekt festgestellt werden könnte.
Die in vielen Publikationen erwähnte verschwindend geringe Prozentzahl der im Screening gefundenen falsch-positiven MCAD-Verdachtsfälle rührt daher, dass diese Fälle in Statistiken meist in Relation zur Gesamtanzahl aller gescreenten Neugeborenen gesetzt werden und nicht nur zu denjenigen, bei denen das Screening den Verdacht auf einen MCAD-Mangel überhaupt erst hat aufkommen lassen. Wie bei allen Statistiken muss man immer ganz genau darauf achten, was die betreffende Aufstellung ausdrücken soll. Die Zuverlässigkeit des Screening ist insgesamt sehr groß, denn es werden bei ca 700.000 pro Jahr gescreenten Kindern weniger als 0,02% falsch-positive MCAD-Verdachtsfälle aufgestellt. Dies bedeutet aber nicht, dass ein aufgekommener MCAD-Verdacht mit fast 100%iger Wahrscheinlichkeit zutreffend ist, sondern dass bei “nur” etwa 140 dieser 700.000 Kinder (0,02%) der MCAD-Verdacht fälschlicherweise gemeldet wurde. Unter Berücksichtigung der Tatsache, dass das NG-Screening insgesamt aber ohnehin nur etwa 200 MCAD-Verdachtsfälle (ca 0,03%) aufgebracht hat, ist auf diese bezogen die Anzahl der falsch-positiven Fälle nun doch wieder als sehr groß zu betrachten.
Klassifizierung des Screeningergebnisses
Genaugenommen liefert die Analyse des Acylcarnitinprofils erst einmal nur eine Reihe von Zahlen, die in ihrer Gesamtheit interpretiert werden müssen. Wegweisend für die Diagnose eines MCAD-Mangels ist zwar ein deutlich erhöhter C8-Wert, jedoch existieren dafür möglicherweise auch andere Gründe. Z.B. kann es auch bei völlig MCAD-freien Frühgeborenen im Zuge der künstlichen Ernährung zu einem Ansteigen des C8-Levels kommen, da die speziell auf ihre Bedürfnisse zugeschnittene Nahrung oftmals einen hohen Anteil mittelkettiger Fettsäuren (MCT-Fette) enthält.
Die sichere Feststellung, ob ein Kind nun tatsächlich einen MCAD-Mangel aufweist, nur Carrier ist, oder die auffälligen Werte eine andere Ursache haben könnten, ist eine komplizierte Angelegenheit. Die Spannbreite der bisher im NG-Screening gefundenen auffälligen C8-Werte ist auch bei gleichen genetischen Mutationen sehr groß und zwischen den verschiedenen Gruppen, in die man die verschiedenen Fälle unterteilen könnte, gibt es gewisse Überlappungsbereiche. Damit ist sowohl die Unterscheidung zwischen normalen und milden MCAD-Ausprägungen, als auch zwischen milden Ausprägungen und Carrieren oder überhaupt nicht Betroffenen, die nur aufgrund der MCT-haltigen Ernährung auffällig Werte zeigen, oft sehr schwer.
Schon seit einigen Jahren suchen und testen die Wissenschaftler aus dem Acylcarnitinprofil ableitbare Merkmale, anhand derer sich ein MCAD-Mangel bereits im NG-Screening möglichst früh und sicher erkennen lassten sollte. Da erhöhte Level bei C6, C8, C10 usw. unter Umständen auch aus ganz anderen Gründen auftreten und im Zuge der regelmäßigen Nahrungsaufnahme nach einigen Tagen auch bei Vorliegen eines MCAD-Mangels deutlich absinken können, greift man dabei vor allem auf Verhältniswerte zwischen einzelnen Acylcarnitinkonzentrationen zurück und hofft, bei der Eintragung dieser Quotienten in ein Diagramm bestimmte Muster auszumachen, die jederzeit eine möglichst sichere Klassifizierung ermöglichen. Anhand einer größeren Anzahl untersuchter MCAD-Fälle mit nachgewiesenen genetischen Mutationen kann man dann beispielsweise ein solches Diagramm aufstellen:
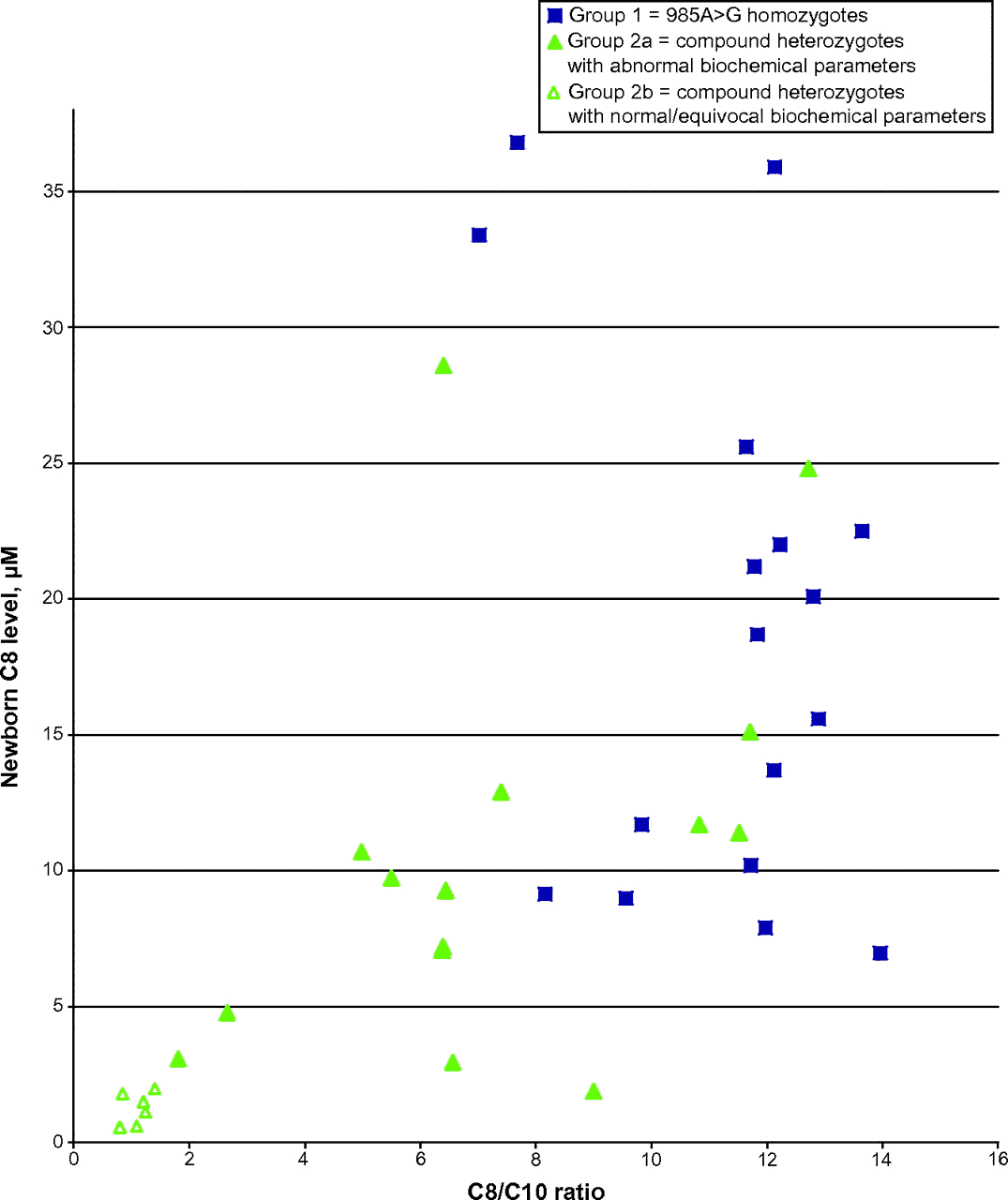
Anhand dieses Diagramms fällt dann z.B. auf, dass sich alle untersuchten Fälle mit zwar compound heterozygoten Mutationen, aber gleichzeitig nur gering erhöhten C8-Werten (offene Dreiecke), in Kombination mit dem C8/C10-Quotienten zu einer Wolke im linken unteren Bereich formen, während die nachgewiesenen K329E homozygot-Fälle (blaue Quadrate) auch bei relativ niedrigen C8-Werten ein dennoch großes C8/C10-Verhältnis aufweisen. Die dazwischen liegenden compound heterozygoten Varianten mit deutlich auffälligen biochemischen Werten (geschlossene Dreiecke) können anhand dieses Diagramms je nach Lage mehr den bekannten Risikovarianten oder den vermutlich milderen Ausprägungen zugerechnet werden.
Während das C8-Level alleine noch keine Klassifizierung zulässt, kann man unter Hinzunahme des C8/C10-Quotienten eine genauere Unterteilung vornehmen. Andere Verhältniswerte werden ebenfalls untersucht und verwendet, jedoch scheint hinsichtlich der Diagnose des MCAD-Mangels − insbesondere der riskanteren Varianten − hauptsächlich C8/C10 hilfreich zu sein, die anderen Quotienten können dem Experten aber in den trotzdem noch möglichen Grenzfällen zur weiteren Absicherung der Diagnose dienen.
Vorsicht bei Fastentests!
Gerade dann, wenn wie beim MCAD-Mangel eine Krankheit nur aufgrund irgendwelcher auffälliger Laborwerte diagnostiziert wird und sich bei dem Kind keinerlei äusserlich sichtbare Auffälligkeiten finden lassen, fällt es den Betroffenen verständlicherweise zunächst sehr schwer, auch tatsächlich an diesen Befund zu glauben, denn der hoffnungsvolle Gedanke “vielleicht wirkt es sich bei meinem Kind ja ganz anders aus” bleibt lange, wenn nicht gar für immer bestehen. Oft ist die Abklärung des Befundes in der Klinik auch nicht so umfangreich und hieb- und stichfest, wie man es sich als Eltern wünscht. Trotzdem sollte an den “Extremtest”, also das Kind gezielt hungern zu lassen, um zu beobachten, ob und was mit ihm nach einigen Stunden passiert, nicht einmal gedacht werden! Wenn überhaupt, liese sich das nur unter ärztlicher Aufsicht in der Stoffwechselklinik mit vollständig vorbereiteten Notfallmaßnahmen durchführen und selbst damit könnte nicht hundertprozentig garantiert werden, dass die Stoffwechselentgleisung am Ende noch rechtzeitig gestoppt und das Kind gerettet werden könnte − und dies, ohne dass es bleibende Schäden davonträgt.
Eine wissenschaftliche Veröffentlichung aus dem Jahr 2002 (Fatty acid oxidation disorders, P.Rinaldo, D.Matern) fordert als sicheren Beweis eines milden MCAD-Mangels zuerst die Feststellung einer normalen klinischen und labormedizinischen Reaktion auf langes Fasten von mindestens 24 Stunden. Erst dann könne bei dem betreffenden Patient, bzw. der vorliegenden Mutationskombination, von einer milden Variante ausgegangen werden. Da man jedoch nie ausschließen kann, dass ein solcher Versuch nicht doch in einer akuten metabolischen Krise endet, verzichten die meisten Ärzte trotz des vorliegenden Verdachts einer milden Variante auf eine entsprechende weiterführende Abklärung der Diagnose und behandeln diese Patienten wie bei Vorliegen eines normalen MCAD-Mangels, selbst wenn es bei ihnen auch ohne Behandlung vielleicht niemals zu einer Stoffwechselentgleisung kommen würde.
Als Mittel zur Feststellung, ob bei einem Kind tatsächlich ein MCAD-Mangel vorliegt − oder vielleicht doch keiner? − ist dieses Fasten-Verfahren strikt abzulehnen und wird vermutlich auch von keinem Arzt in Erwägung gezogen.
Trotzdem kann es vorkommen, dass den Eltern eines vom MCAD-Mangel betroffnen Kindes von ihrer Stoffwechselambulanz ein Austesten der noch “sicheren” maximalen Mahlzeitenabstände vorgeschlagen wird. Mittels kontinuierlicher Kontrolle der Blutzuckerkonzentration und sichtbarer klinischer Symptome soll dabei festgestellt werden, nach welcher Zeitspanne ohne Nahrungsaufnahme die Zuckerreserven der Leber aufgebraucht sind und der Blutzuckerspiegel in kritischer Weise abfällt. Eine entsprechende niederländische Studie an MCAD-Patienten im Alter von 2,5 Monaten bis 20 Jahren hat gezeigt, dass sich in rund 20% der Tests klinische Symptome zeigten, noch bevor eine Unterzuckerung festgestellt werden konnte. Dies läßt sich damit erklären, dass der Körper bereits zu einem wesentlich früheren Zeitpunkt mit der Mobilisierung der Fettreserven beginnt, um die vollständige Entleerung der Glycogenvorräte zu verhindern, oder wenigstens möglichst weit hinauszuzögern. Während einer längeren Hungerphase ist ein normaler Blutzuckerspiegel folglich kein Beweis dafür, dass noch alles “im grünen Bereich” ist, denn es kann sich schon längst eine katabole Stoffwechsellage eingestellt haben. Eltern, denen solch ein Austesten empfohlen wird, sollten daran denken, dass sie diesem Vorschlag keineswegs zustimmen müssen, und sie selbst es sind, die letztlich die Verantwortung für ihr Kind und die eventuellen Folgen eines solchen Versuchs zu tragen haben werden.
Auch dient so ein Fastentest mehr zur Generierung statistischer Daten, als zur tatsächlichen Feststellung, wie lange die Nahrungspausen bei einem Kind wirklich sein dürfen. Denn auch wenn während eines unter ärztlicher Aufsicht stattgefundenen Tests eine verlängerte Nüchternphase von z.B. 10 Stunden, statt der möglicherweise noch empfohlenen 8 Stunden ohne Auswirkungen blieb, bedeutet dies nicht, dass man sich auf dieses Einzelergebnis grundsätzlich verlassen kann. Wie lange die im Körper vorhandenen Kohlenhydrate tatsächlich ausreichen, hängt von vielen, zahlenmäßig nicht genau erfassbaren Faktoren ab, z.B. wieviel Kohlenhydrate das Kind bei der letzten Mahlzeit zu sich genommen hat, ob es während der betreffenden Zeitspanne aktiv ist oder schläft, und ob es gesund ist, oder ob sein Körper einen mehr oder weniger starken Infekt bekämpfen muss, der vielleicht noch gar nicht als solcher deutlich erkennbar ist.
Die Aussagekraft von Blutzuckermessungen beim MCAD-Mangel
Das Hauptrisiko beim MCAD-Mangel besteht zwar aus der sich aufgrund einer massiven Unterzuckerung entwickelnden Unterversorgung der wichtigsten Organe, aber aus den im letzten Abschnitt genannten Gründen ist auch die regelmäßige tägliche Kontrolle des Blutzuckerspiegels eine völlig überflüssige Maßnahme und führt zu nichts − ausser zu einer erhöhten Stressbelastung des Kindes und der Eltern. Zeigt ein Kind jedoch plötzlich ein auffälliges und besorgniserregendes Verhalten, sollten die Eltern trotzdem sofort den Blutzucker kontrollieren und zu diesem Zweck bereits über ein entsprechendes Gerät verfügen, welches von der Stoffwechselambulanz oder vom Kinderarzt verschrieben werden kann und von einem Fachmann (z.B. in einer Apotheke) für die korrekte Anzeige niedriger Blutzuckerwerte geeicht werden muss − standardmäßig sind solche Geräte nämlich für die zuverlässige Ermittlung zu hoher Blutzuckerwerte bei Diabetikern eingestellt und im niedrigen Bereich ungenau. Ein bereits deutlich erniedrigter Blutzuckerspiegel wäre dann als Hinweis auf einen absoluten Notfall und höchste Eile zu werten, so dass die Eltern sich nicht mal mehr selbst mit dem Kind auf den Weg in die Klinik begeben, sondern besser sofort den Notruf 112 wählen sollten.
Doch auch bei einem unauffälligen Messergebnis sollte man folgendes unbedingt beachten: der Blutzuckerspiegel kann bei der Messung völlig normal erscheinen, obwohl die Entgleisung schon in vollem Gange ist und das Kind bereits entsprechende klinische Symptome, wie z.B. Apathie zeigt. Als Eltern muss man in einer solchen Situation unbedingt auf das eigene Bauchgefühl hören und darf sich nicht auf Apparate verlassen. Das Blutzuckermessgerät kann eine laufende Entgleisung zwar eventuell beweisen, jedoch niemals widerlegen! Die heimische Messung des Zuckerwertes erlaubt keine verlässliche Aussage hinsichtlich des tatsächlichen metabolischen Status des Kindes und führt aufgrund der u.U. zum Zeitpunkt der Messung noch normal erscheinenden Werte möglicherweise sogar dazu, dass man sich viel zu spät auf den Weg ins Krankenhaus macht. Umgekehrt können bei regelmäßiger täglicher Kontrolle aber auch ganz normale und unkritische Schwankungen des Blutzuckerspiegels schon zu starker Besorgnis der Eltern und somit zu einem ständig erhöhten, aber völlig unnötigen Stresslevel führen.
Ein Mitglied hat im Forum von dem negativ zu bewertenden Erlebnis berichtet, dass sie beim ersten Besuch ihrer nächstgelegenen Stoffwechselambulanz lediglich mit einer Verschreibung für Carnitin-Sirup und für ein Blutzuckermessgerät (verbunden mit der Anweisung täglich mehrmals zu messen), aber ohne jegliche weitere Untersuchungen zur Abklärung der Diagnose nach Hause geschickt wurde. In solchen Situationen, in denen man als Eltern zu Recht das Gefühl hat, weitgehend im Regen stehen gelassen zu werden, sollte man sich nicht scheuen, zukünftig auch den längeren Weg zu einem anderen Stoffwechselzentrum auf sich zu nehmen. Da sich die regelmäßigen Untersuchungen und die Notwendigkeit der kurzfristigen Behandlungen in schwierigen Krankheitssituationen über viele Jahre erstrecken werden, sollten die Eltern ihrem zuständigen Arzt gegenüber Vertrauen haben können, oder zumindest überzeugt sein, dass er bzgl. des MCAD-Mangels weiß wovon er redet.
Die molekulargenetische Analyse
Wurde der Verdacht auf einen MCAD-Mangel durch ein zweites Screening erhärtet, erfolgt üblicherweise eine Untersuchung des Erbguts hinsichtlich einer Mutation im zuständigen ACADM-Gen. Wen die genaue Funktionsweise der Proteinsynthese anhand des genetischen Codes der DNA interessiert und was eine Mutation innerhalb eines Nukleobasen-Tripletts dabei anrichtet, kann es im Artikel “Infos zu bekannten Mutationen” nachlesen. An dieser Stelle würde das Thema jedoch zu weit führen.
Ganz simpel ausgedrückt, muss man sich aber ein Chromosom als einen in lauter Schleifen verschlungenen DNA-Faden vorstellen. Dieser Faden ist eine Art langes Computerprogramm, welches in seinen einzelnen Abschnitten (den Genen) den notwendigen Programmcode zur Herstellung verschiedener Bausteine, z.B. von Enzymen enthält. Ein Computerprogramm macht allerdings nur dann genau das, was es soll, wenn sein Programmcode von der ersten bis zur letzten Zeile fehlerfrei durchlaufen werden kann. Ein Programmierfehler innerhalb dieses Codes führt zu einem der allseits bekannten und ungeliebten Abstürze. Genau so verhält es sich auch mit dem Gen, welches für die Bildung des MCAD-Enzyms zuständig ist. Durch eine Mutation auf molekularer Ebene wird gewissermaßen ein einziges falsches Zeichen in den betreffenden Programmcode eingeschmuggelt, welches entweder zum vollständigen Absturz führt, so dass der Körper nicht in der Lage ist, das Enzym zu bilden, oder aber in der Bildung eines nicht voll funktionsfähigen MCAD-Enzyms resultiert. Dabei ist es relativ egal, an welcher Stelle der Fehler in den Programmcode eingebaut wird − er führt in jedem Fall zum Absturz oder zum Fehler. Dies erklärt, weshalb inzwischen eine ganze Reihe unterschiedlicher Mutationen als Verursacher des MCAD-Mangels identifiziert wurden − auch wenn es sich in den meisten Fällen um die bekannte K329E Mutation handelt. Allerdings läßt sich auch bei Patienten mit K329E homozygot meist noch eine geringe − und für den MCAD-Mangel typische − Enzymaktivität feststellen, die jedoch im Ernstfall nicht ausreicht, um das Gehirn während einer längeren Hungerphase mit der benötigten Energie zu versorgen.
Das ACADM-Gen besteht aus 12 Exons, die häufigste Mutation K329E befindet sich an der Position 985 in Exon 11 (daher auch die Bezeichnung 985A>G) und besteht in einer Veränderung von Adenin zu Guanin, welche im MCAD-Enzym einen Aminosäurenaustausch von Lysin zu Glutamat und damit einen drastischen Einbruch der Enzymaktivität bewirkt. Üblicherweise starten die Labore die Sequenzierung des Gens daher in Exon 11. Nur wenn an dieser bekannten Stelle keine, oder nur eine heterozygote Mutation existiert, wird die Suche auf alle restlichen Exons ausgedehnt.
Nach Auskunft eines Screeningzentrums wird für gesetzlich krankenversicherte Patienten generell das gesamte Gen sequenziert. Bei privat versicherten Patienten dagegen beschränkt sich die molekulargenetische Untersuchung anscheinend zunächst auf die Suche nach der häufigsten Mutation in Exon 11. Soll das vollständige Gen sequenziert werden, ist dafür die Zustimmung des das Kind betreuenden Arztes erforderlich, da gegenüber vielen privaten Versicherungen zwecks Kostenübernahme die unbedingte Notwendigkeit einer solchen Untersuchung begründet werden muss. Sehen Arzt oder in zweiter Instanz die Krankenkasse dies nicht als notwendig an, kann es sein, dass die Suche nach den verantwortlichen Mutationen auf Exon 11 beschränkt bleibt und im Ergebnis unter Umständen keine, oder zumindest keine zweite für den vermuteten MCAD-Mangel verantwortliche Genmutation aufgespürt wird.
Sollte das Screening nur grenzwertige Acylcarnitinwerte aufgewiesen haben, sollte man sich als Eltern in so einem Fall genau überlegen, ob man sich mit der dann oft trotzdem gestellten Diagnose MCAD-Mangel abfindet, oder entsprechende Anstrengungen für eine weitergehende Abklärung unternimmt.
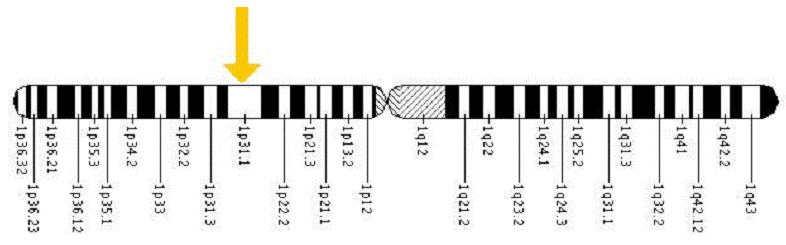
Um noch eine genauere Lagebeschreibung zu liefern: das ACADM-Gen selbst befindet sich auf dem kurzen Arm von Chromosom 1 an Position 31, und umfasst die Basenpaare 75.962.869 bis 76.001.770. Es hat somit eine Länge von fast 39.000 Basenpaaren, in denen bei der molekulargenetischen Untersuchung der eine Codierungsfehler gefunden werden muss, der für die Fehlbildung des MCAD-Enzyms verantwortlich ist. Allerdings haben nur 1263 dieser Basen eine codierende Bedeutung. Alle anderen befinden sich auf Introns, die während der zur Bildung des Emzyms ablaufenden “Translation” aus dem Code entfernt werden oder auf sonstigen nicht-codierenden Bereichen. Trotzdem können auch auf diesen Introns liegende Mutationen unter gewissen Umständen negative Auswirkungen auf die korrekte Enzymbildung haben.
An Position 199 in Exon 3 liegt eine weitere häufige Mutation (199T>C), die einen Austausch von Tyrosin zu Histidin bewirkt und für die etwa jeder 500. Mensch Carrier ist. Diese und viele weitere Mutation wurden bisher jedoch ausschließlich bei im NG-Screening (leicht) auffälligen Kindern gefunden und nie bei Patienten mit erfolgter Stoffwechselentgleisung.
Weitere Untersuchungen anhand von Blut und Urin
Neben der molekulargenetischen Untersuchung kann der Arzt noch weitere Blut- und Urinanalysen veranlassen. Leider hat sich auch dabei noch kein einheitliches Verfahren etabliert und von Stoffwechselzentrum zu Stoffwechselzentrum unterscheidet sich stark, welche Untersuchungen als sinnvoll erachtet und durchgeführt werden. Ein Arzt gibt sich mit einer gefundenen Genmutation zufrieden und verzichtet auf jede weitere Untersuchung, da sie ihm überflüssig erscheinen. Ein anderer Arzt läßt aus Blut und Urin eine Reihe weiterer Parameter bestimmen, um sich ein umfassenderes Bild über das zu erwartende Ausmaß einer möglichen Stoffwechselentgleisung machen zu können. Zum Beispiel werden dabei der Blutzucker, der Säure-Basen-Status, die Leberfermente und eine Reihe weiterer Blutwerte bestimmt. Die Untersuchung der organischen Säuren im Urin ist diagnostisch nur sehr eingeschränkt nützlich. Dabei findet man unter Umständen − nicht immer − eine erhöhte Konzentration von Adipinsäure, Suberin- und Sebacinsäure sowie der Glycinkonjugate Hexanoylglycin und Suberylglycin. Die alleinige Untersuchung der organischen Säuren kann somit nicht als Bestätigungsdiagnostik dienen, jedoch liefert sie u.U. ein weiteres ins Bild passendes Puzzleteil.
Anmerkung:
Hinsichtlich der Aussagekraft und Verlässlichkeit dieser zusätzlichen Untersuchungen gibt es unterschiedliche Auffassungen. Ursprünglich folgte an dieser Stelle der Satz:
“Im Gegenzug kann man aber auch mit einem völlig unauffälligen Befund in diesen zusätzlichen Unterschungen, einen in den Screenings gefundenen und bestätigten MCAD-Mangel nicht mehr diagnostisch ausschließen, denn wenn das Kind zum Zeitpunkt der Blut- und Urinprobe vollkommen gesund ist, also eine ausgeglichene Stoffwechsellage hat, werden auch die Analyseergebnisse völlig normal erscheinen.”
Richtig ist, dass es aufgezeichnete Fälle von (jugendlichen und erwachsenen) MCAD-Patienten gibt, bei denen − trotz bereits nachgewiesener K329E homozygot-Mutation − die Urinuntersuchungen keinerlei erhöhte Konzentrationen der oben genannten organischen Säuren aufzeigten. Insofern taugt ein allein in diesen Untersuchungen ermitteltes unauffälliges Ergebnis nicht, um einen bisher nur rein auf Trockenblut-Screenings basierenden MCAD-Verdacht sicher zu entkräften. Entscheidend ist das Resultat der Enzymaktivitätsuntersuchung, das in Einzelfällen auch noch ggf. durch eine molekulargenetischen Untersuchung unterstützt werden sollte.
Gegen die uneingeschränkte Korrektheit der zweiten Satzhälfte (“…wenn das Kind zum Zeitpunkt der Blut- und Urinprobe vollkommen gesund ist, also eine ausgeglichene Stoffwechsellage hat, werden auch die Analyseergebnisse völlig normal erscheinen.”) sprechen aber die Erfahrungen einiger Forumsteilnehmer, bei deren Kindern mit zweifelsfrei diagnostiziertem MCAD-Mangel bei allen Tests (sowohl den weiteren Trockenblutkarten, als auch den Blut- und Urinuntersuchungen) immer die für den MCAD-Mangel charakteristischen Substanzen in deutlich erhöhter Konzentration gefunden wurden − trotz aktuell ausgeglichener Stoffwechsellage.
Von einigen Ärzten werden auch weitere Kontrollen mittels Trockenblutkarten als unnötig angesehen, da nach ihrer Auffassung bei gut ernährten und gesunden Kindern nur noch unauffällige Screening-Ergebnisse zu erwarten seien und zusätzliche Kontrollen nach den ersten Lebenstagen des Kindes daher keinen Sinn machten.
Gerade bezüglich dieser in Frage gestellten Aussagekraft von Trockenblutkarten nach den ersten Lebenstagen eines Kindes, wurde im Forum von einem Fall berichtet, bei dem ein im NG-Screening wegen leicht erhöhter Werte vermuteter MCAD-Mangel, aufgrund eines erst nach 6 Monaten durchgeführten zweiten Screenings mit unauffälligem Ergebnis mit absoluter Sicherheit ausgeschlossen wurde. Auch ein möglicherweise bereits bei den Eltern vorliegender MCAD-Mangel wird von einigen Stoffwechselambulanzen anhand eines unauffälligen Trockenblutkarten-Befundes diagnostisch ausgeschlossen, während diese Analysemethode in anderen Stoffwechselambulanzen als für Erwachsene untauglich und nicht aussagekräftig angesehen wird.
Vom Screening Zentrum Hessen erfolgte auf eine diesbezüglich Anfrage hin die folgende Antwort:
“MCAD-Defekte lassen sich im Trockenblut nicht nur nach der Geburt nachweisen, sondern auch später. [..] Die charakteristischen Veränderungen der Primärparameter (C8, C6, C10 und C10:1) sowie der Sekundärparameter (C8/C2, C8/C6, C8/C10, C8/C12, C8/16 etc.) lassen sich lebenslang nachweisen.”
In der Monatsschrift “Kinderheilkunde” (2006 · 154:1231–1244, DOI 10.1007/s00112-006-1425-1, Online publiziert: 7. November 2006, Springer Medizin Verlag 2006) wurde diese Problematik im Artikel “Störungen der Fettsäureoxidation” behandelt und folgendermaßen beantwortet:
“Für den VLCAD-Mangel hat sich in einzelnen Fällen gezeigt, dass aufgrund des Anabolismus nach Ablauf der ersten Lebenstage das Acylkarnitinprofil am Ende der ersten Lebenswoche nicht mehr ausreichend diagnostisch wegweisend ist. Dies hat zu einem unauffälligen Zweitscreening trotz Vorliegen eines VLCAD-Mangels geführt. Es ist daher hinsichtlich der Störungen der Fettsäurenoxidation extrem wichtig, das Neugeborenenscreening während des empfohlenen Abnahmezeitpunkts zwischen der 48. und 72. Lebensstunde durchzuführen und nicht später.”
Da der MCAD-Mangel im gleichen Abschnitt wie der VLCAD-Mangel behandelt, aber in diesem Zusammenhang nicht erwähnt wurde, scheint es sich tatsächlich um eine ausschließlich beim VLCAD-Mangel, und auch dort nur sehr selten auftretende Problematik zu handeln, die bei der Diagnostik des MCAD-Mangels bisher noch nicht beobachtet werden konnte.
Wie sieht es mit dem Risiko für die Eltern und die Geschwister aus?
Die Eltern eines vom MCAD-Mangel betroffenen Kindes müssen gemäß der Vererbungslehre beide zwingend zumindest heterozygote Träger (Carrier) eines mutierten Allels sein. Natürlich könnte auch nur ein Elternteil Carrier sein und die Mutation im vom anderen Elternteil beigesteuerten MCAD-Gen des Kindes spontan entstanden sein, aber das passiert so extrem selten, dass es hier nur als hypothetische Möglichkeit angesehen werden soll. Ein solcher Fall ist in zehn Jahren MCAD-Forum unter den sich über die Zeit beteiligenden rund 550 Familien (somit 1100 Elternteilen) auch nie aufgetreten oder zumindest nie bekannt geworden. Es ist somit nicht völlig auszuschliessen, sollte aber aber auch nicht als wahrscheinlich angenommen werden. Üblicherweise werden beide Mutationen von beiden Elternteilen vererbt. Als Carrier sind sie asymptomatisch, haben selbst also keinen MCAD-Mangel und stehen somit auch nicht in der Gefahr, eine Stoffwechselentgleisung zu erleiden.
Trotzdem sollte eine genetische Überprüfung oder zumindest die Prüfung des Acylcarnitinprofils sicherheitshalber auch bei den Eltern erfolgen, denn die von einem Kinderarzt gemachte Aussage: “Dass sie hier vor mir stehen, zeigt bereits, dass Sie selbst keinen MCAD-Mangel haben können, denn sonst hätten Sie vermutlich gar nicht bis jetzt überlebt!” ist zwar aufgrund der tatsächlich extrem geringen Wahrscheinlichkeit für einen solchen Fall schon als beruhigendes Indiz, aber keineswegs als sicherer Beweis zu werten! Bereits seit 1986 gibt es immer wieder medizinische Publikationen, die von zwar asymptomatischen, jedoch homozygoten Elternteilen mit echtem, klassischem MCAD-Mangel berichten. Da vor der Einführung der Tandemmassenspektrometrie nur sehr wenige Mutationen des ACADM-Gens überhaupt bekannt waren, beziehen sich diese Berichte sogar fast immer auf die Hochrisikovariante K329E. Wird diese Mutation bei einem Kind gefunden, sollten die Eltern ebenfalls genau untersucht werden, da in einigen Fällen auch bei wenigstens einem Elternteil ein echter MCAD-Mangel vorliegen könnte, der zum Glück noch nicht in Form einer Entgleisung oder sonstiger Symptome in Erscheinung getreten ist. Natürlich können die Eltern auch andere ACADM-Mutationen homozygot oder compound heterozygot aufweisen.
Auch Geschwisterkinder, die vor der Einführung des erweiteren Neugeborenenscreenings auf die Welt kamen und bisher symptomfrei blieben, müssen unbedingt hinsichtlich eines möglichen MCAD-Mangels untersucht werden. Für sie besteht ebenfalls ein 25%-Risiko, von ihren Eltern jeweils das Chromosom mit dem defekten Gen geerbt zu haben. Mit einer Wahrscheinlichkeit von 50% sind sie nur asymptomatische Carrier, doch auch das sollten sie im Hinblick auf ihre zukünftigen eigenen Kinder wissen. Die Kontrolle der Geschwister sollte auch dann stattfinden, wenn sich herausstellt, dass das gescreente Neugeborene doch nur einen heterozygoten Defekt hat, und lediglich Carrier ist.
Problematisch und fast unmöglich zu entdecken sind natürlich die Fälle, in denen ein seit etwa 2005 geborenes Kind die beiden normalen Allele seiner Eltern abbekommen hat und im Screening somit kein MCAD-Defekt nachweisbar war, sein älteres Geschwisterkind jedoch vor der Einführung des erweiterten NG-Screening geboren wurde und sein echter MCAD-Mangel daher noch unentdeckt blieb. Das betrifft heute (2022) also vor allem junge Erwachsene. In den Erfahrungsberichten auf der amerikanischen Selbsthilfe-Website www.fodsupport.org rufen daher viele Eltern dazu auf, unbedingt auch ältere Kinder notfalls auf eigene Kosten per Screening nachtesten zu lassen, da der MCAD-Mangel ihrer eigenen Kinder erst entdeckt wurde, als es schon zu spät war. Niemand von ihnen hatte zuvor irgendetwas geahnt, geschweige denn etwas vom MCAD-Mangel gehört. Die derart betroffenen jungen Erwachsenen stehen allerdings vor einem nicht unbeträchtlichen Problem, da sie nicht mehr in die Zuständigkeit der Kinderärzte und der pädiatrischen Abteilungen der Uni-Kliniken fallen, sondern mit Ärzten und Kliniken zu tun bekommen werden, die auf Erwachsenenmedizin spezialisiert sind und daher immer noch über so gut wie keine Kenntnisse zum MCAD-Mangel verfügen und entsprechend auch nicht zuerst in diese Richtung denken, wenn ein Erwachsener mit entsprechenden Symptomen, aber ohne Wissen um seinen MCAD-Mangel, zu ihnen kommt.
Kostenübernahme der genetischen Untersuchung der Eltern durch die Krankenkassen
Gerade was die Durchführung der oben empfohlenen molekulargenetischen Untersuchung der Eltern eines Kindes mit MCAD betrifft, gibt es innerhalb der deutschen Uni-Kliniken zwei völlig konträre Ansichten. Während einige Einrichtungen diese für die weitere Diagnose eventuell wichtige Untersuchung den Eltern generell vorschlagen, wird sie in anderen Kliniken mit der Begründung abgetan, der damit verbundene Aufwand und die Kosten seien zu hoch und schließlich ändere sich damit auch nichts mehr am MCAD-Befund des Kindes. Daher sei die elterliche Genanalyse absolut nicht notwendig und würde folglich von den Krankenkassen auch nicht bezahlt.
Diese Aussage ist jedoch nicht zutreffend! Grundsätzlich gilt für alle monogenetisch (= nur von einem einzigen Gen ausgehend) bedingten Erbkrankheiten, zu denen auch der MCAD-Mangel gehört, dass sowohl die molekulargenetische Untersuchung des Kindes, als auch die der Eltern, ein gängiger und ganz selbstverständlicher Diagnoseschritt ist, der daher von dem behandelnden Arzt auch sehr einfach und plausibel gegenüber den Krankenkassen begründbar ist und von diesen bei Vorlage einer solchen ärztlichen Begründung üblicherweise völlig problemlos übernommen wird.
Ein wesentliches Argument ist beispielsweise, dass man im Hinblick auf eventuell noch folgende Kinder wissen sollte, wie groß deren Risiko ist, ebenfalls den MCAD-Mangel zu erben, damit sich die Eltern und das Krankenhauspersonal darauf einrichten können, um bereits für die ersten Tage nach der Geburt alle notwendigen Maßnahmen zur Sicherstellung einer ausreichenden Ernährung zu ergreifen. Gerade bei compound heterozygoten Mutationen ergibt die Untersuchung der Eltern nämlich möglicherweise, dass das Kind den Gendefekt nur von einem Elternteil geerbt und den MCAD-Mangel erst aufgrund einer zufälligen spontanen Mutation auf der zweiten Genkopie ausgebildet hat. Die Gefahr, dass sich so ein Fall wiederholt, wäre somit sehr gering.
Zudem könnten sich Paare mit stark erhöhtem Risiko der Weitervererbung (wenn z.B. ein Elternteil selbst homozygot ist) dazu entschließen, die Familienplanung vorzeitig abzuschliessen. Auch wenn das Risiko der erneuten Vererbung des MCAD-Mangels keinen Grund darstellen sollte, auf weitere Kinder zu verzichten, wäre dies auf jeden Fall ein schlüssiges und unwiderlegbares Argument zur Begründung der Notwendigkeit der genetischen Untersuchung der Eltern.
Sollte der behandelnde Arzt trotzdem auf einer ablehnenden Haltung beharren, könnte es angebracht sein, bei einer anderen Stoffwechselklinik eine zweite Meinung einzuholen, bzw. dort um eine diesbezügliche weitere Abklärung zu ersuchen.
Die Entstehungsgeschichte des erweiterten Neugeborenenscreenings
Die Bezeichnung “erweitertes” Neugeborenenscreening rührt übrigens daher, dass zwar schon seit längerem ein generelles Neugeborenenscreening durchgeführt wird, eine ganze Reihe seltener Stoffwechselstörungen, wie z.B. auch der MCAD-Mangel, darin aber noch nicht enthalten waren, da man sich zunächst hauptsächlich auf die Früherkennung von Krankheiten mit einem sehr schweren Krankheitsbild beschränkte. Nach und nach begannen die Screeningzentren einzelner Bundessländer den Nutzen eines um eine Reihe weiterer Stoffwechselstörungen erweiterten Screenings zu untersuchen. Bayern spielte dabei bereits seit 1999 mit einem international beachteten Modellprojekt eine Vorreiterrolle. Weitere Bundesländer schlossen sich in den folgenden Jahren diesem Beispiel an. Allerdings war das erweiterte Screening bis 2005 noch keine generelle Kassenleistung, sondern wurde nur von einigen gesetzlichen Krankenkassen übernommen. Bis dahin bekamen die Eltern bei der Geburt ihres Kindes das erweiterte Screening neben dem Standardscreening zwar von Jahr zu Jahr in immer mehr Bundesländern angeboten, mussten dessen Kosten (ein Bericht zum NG-Screening nennt hier eine Betrag von ca. 20€) aber u.U. selbst tragen, so dass in einigen Fällen die Entscheidung möglicherweise auch gegen dieses Angebot ausfiel.
Seit 2005 ist das erweiterte Screening endlich in ganz Deutschland Regelleistung der gesetzlichen Krankenkassen. Damit war Deutschland das weltweit erste Land, welches das erweiterte Screening anhand der Tandem-Massenspektrometrie zum gesetzlichen Versorgungsstandard machte. Die Durchführung des Screenings erfordert aber nach wie vor die unterschriebene Einverständniserklärung der Eltern, und leider lehnen immer noch einige Eltern diese Untersuchung aus den verschiedensten Gründen ab. Eine Familie in unserer Bekanntschaft erwähnte uns gegenüber mal, dass sie ihr jüngstes Kind aufgrund unseres Erlebnisses mit dem uns über mehrere Monate beunruhigenden falschen Alarm, absichtlich nicht mehr hätten screenen lassen. Lieber gingen sie das Risiko ein, dass ihr Kind dann doch etwas habe und sie es nicht wüssten, als dass es nichts habe, sie aber falsch informiert und dadurch beunruhigt würden. Wir konnten diese Sichtweise damals absolut nicht nachvollziehen!
Seit der bundesweiten Einführung des erweiterten Neugeborenenscreenings im Jahr 2003 werden statistische Daten über die Häufigkeit der zusätzlich geprüften Stoffwechselstörungen erhoben. Kinder mit angeborenem MCAD-Mangel bilden unter den statistisch erfassten Neugeborenen mit Enzymdefekten der Fettsäurenoxidation noch die größte Gruppe. Als Beispiel sei das Jahr 2004 genannt, in dem 705.622 Kinder geboren und gescreent wurden. Darunter wurden 72 Kinder gefunden, die eine dieser getesteten Fettsäurenoxidations-Störungen aufwiesen. Mehr als die Hälfte dieser Kinder, nämlich 47, wiesen einen MCAD-Mangel auf. In Bezug auf die Gesamtanzahl der geborenen Kinder ergab sich für 2004 somit eine relative Häufigkeit von etwa 1:15000 [Quelle: Monatsschrift Kinderheilkunde 2006, Springer Medizin Verlag]. Um einen Vergleich zu früher aufzustellen: Vor der Einführung des erweiterten Neugeborenenscreenings wäre ein MCAD-Mangel nur bei 16 bis 20 dieser 47 Kinder gefunden worden − und dann frühestens zu dem Zeitpunkt, wenn sie mit schwersten Krankheitssymptomen auf der Notfallstation des Krankenhauses eingeliefert worden wären.
Wie sind die Prozentangaben für die Häufigkeiten verschiedener Mutationen zu verstehen?
An dieser Stelle möchte ich die Prozentangaben für die Häufigkeit des Auftretens bestimmter bekannter Mutationen noch einmal genauer beleuchten, da sich diese Angaben für die bei der Geburt gescreenten Kinder und diejenigen Kinder, deren MCAD-Mangel erst nach einer ersten Stoffwechselentgleisung festgestellt wurde, drastisch unterscheiden. In medizinischen Publikationen ist häufig zu lesen, dass die prävalente Mutation K329E (985A>G) homozygot in rund 80% aller Fälle vorliegt. Dieser Wert stammt jedoch aus der Zeit vor der Aufnahme des MCAD-Mangels in das Neugeborenenscreening. Die Zahl wurde anhand der Fälle bestimmt, bei denen der MCAD-Mangel erst nach einer akuten Stoffwechselentgleisung diagnostiziert wurde und bezieht sich somit ausschließlich auf Kinder, die nicht frühzeitig gescreent wurden. Gleiches gilt für die Aussage, dass in 18% der weiteren Fälle K329E compound heterozygot mit einer anderen Mutation als Verursacher des MCAD-Mangels identifiziert wurde.
Bezogen auf die Gesamtheit der gescreenten Kinder, bei denen ein MCAD-Mangel bereits kurz nach der Geburt gefunden wurde, liegt der Anteil von K329E homozygot nur bei etwa 35%, wie eine bayerische Langzeitstudie über 5 Jahre gezeigt hat. Somit läßt sich bereits ein deutlicher Rückschluss auf den Nutzen des erweiterten Neugeborenenscreenings ziehen. Denn nur etwa ein Drittel aller vom MCAD-Mangel betroffenen Kinder weist die Mutation K329E homozygot auf, aber in fast allen früher erfassten Fällen, bei denen es aufgrund eines unbekannten MCAD-Mangels zu einer oftmals sehr folgenschweren Stoffwechselentgleisung kam, war diese Mutation homozygot oder compound heterozygot beteiligt. Man findet heutzutage also noch eine Menge anderer ACADM-Mutationen, die einen nachweisbaren MCAD-Mangel verursachen, gleichzeitig in den meisten Fällen aber einen milden, bzw. milderen Verlauf vermuten lassen, da sie noch nie im Zusammenhang mit Stoffwechselkrisen in Erscheinung traten. Die Langzeitstudie ergab, dass durch die frühzeitige Screening-Diagnose mit darauf folgender entsprechender Behandlung und Begleitung durch Stoffwechselexperten und dank der gleichzeitig erhöhten Wachsamkeit der Eltern im Krankheitsfall des Kindes, das Auftreten einer Stoffwechselentgleisung bisher in fast allen Fällen verhindert werden konnte.
Ich möchte an dieser Stelle noch einmal ausdrücklich darauf hinweisen, dass ich als Autor dieser Seiten größten Wert auf Korrektheit gelegt habe, aber ansonsten medizinischer Laie bin. Alle veröffentlichten Informationen wurden von mir anhand verschiedener verfügbarer Promotionen, Studienarbeiten, medizinischen Veröffentlichungen, MCAD-Broschüren, Merkblätter, Webseiten von Kliniken, Aussagen der Stoffwechselambulanz und sonstigen Erkenntnissen und Erfahrungen (eigenen und denen unserer Forumsteilnehmer) zusammengetragen und in inzwischen weit über 1000 Stunden Arbeitszeit gelesen, unzählige Male überdacht, sortiert und schließlich in der jetzigen Form hier niedergeschrieben. Das Lesen dieser Seiten darf aber auf keinen Fall den Besuch beim Arzt oder der Stoffwechselambulanz ersetzen, sondern soll lediglich dazu dienen, schon mal etwas besser über die ganze Thematik und Problematik Bescheid zu wissen!