Der MCAD-Mangel beeinträchtigt den Stoffwechsel
Der MCAD-Mangel verursacht eine Störung im Fett-Stoffwechsel. Um zu verstehen, was dabei genau gestört ist, soll hier zunächst darauf eingegangen werden, wie der menschliche Stoffwechsel funktioniert, bzw. was das überhaupt ist.
Als Stoffwechsel bezeichnet man die Verarbeitung und Verwertung von Nährstoffen im menschlichen Körper, sowie die damit verbundenen Aufbau- und Abbauvorgänge. Normalerweise funktionieren diese Vorgänge automatisch und völlig reibungslos, solange man sich einigermaßen gesund und ausgewogen ernährt. Bei Menschen mit einer der im Allgemeinen nur selten vorkommenden Stoffwechselstörungen, kann es aber Situationen geben, in denen das normalerweise sehr stabile Gefüge aus dem Gleichgewicht, bzw. aus der Bahn gerät und gewissermaßen “entgleist”.
Ebenso, wie ein entgleister Zug nicht in der Lage ist, wieder von alleine auf die Schienen zurück zu gelangen und seine Fahrt fortzusetzen, können sich auch die Stoffwechselvorgänge im menschlichen Körper während einer laufenden Entgleisung nicht mehr selbst zu einem stabilen Zustand hin regulieren, sondern es müssen von aussen geeignete Gegenmaßnahmen ergriffen werden. Zu diesem Zweck sollten die Eltern, und später auch die vom MCAD-Mangel betroffenen Kinder, wenigstens in Grundzügen über die Stoffwechselvorgänge im Körper Bescheid wissen, denn dann lassen sich problematische Situationen mitunter schon im Vorfeld erkennen und wirkungsvoll vermeiden.
Was ist der Stoffwechsel und wie funktioniert er?
Jedes Lebensmittel, bzw. jede Mahlzeit enthält in unterschiedlichen Mengenanteilen die gleichen Nährstoffe:
- Kohlenhydrate, die z.B. in Form von Zucker oder Stärke vorliegen
- Fette, z.B. in Form von Öl
- Eiweiß (“Proteine”), die besonders in Fleisch, Fisch, Milchprodukten und Eiern in großen Mengen enthalten sind
- Mikronährstoffe, dazu gehören Mineralstoffe, Spurenelemente und Vitamine
- Wasser
Alle diese Stoffe werden während der Verdauung im Magen und im Darm verarbeitet und zu immer kleineren Bestandteilen zerlegt.
Aus Eiweiß werden die Aminosäuren, die das Baumaterial des Körpers bilden. Die Mikronährstoffe haben Helferfunktionen bei allen Vorgängen im Körper. Kohlenhydrate werden zu verschiedenen Zuckern verarbeitet und aus Fetten werden Glycerin und Fettsäuren.
Zucker und Fettsäuren dienen den Organen und besonders dem Gehirn unmittelbar als Energiequellen. Kommt es durch zu reichliche Nahrungsaufnahme aber zu einem Überangebot an Energielieferanten, werden diese für möglicherweise zukünftig folgende schlechte Zeiten aufbewahrt und in Form von Fett im Unterhautfettgewebe und teilweise auch in den Organen eingelagert. Es bilden sich die allseits so unbeliebten Pölsterchen.
Anabolismus und Katabolismus
An dieser Stelle müssen zwei Begriffe eingeführt werden, von denen im Zusammenhang mit dem MCAD-Mangel immer wieder mal die Rede ist. Als Anabolismus bezeichnet man die Phase, während der sich der Körper im Aufbau befindet. Dies ist bei Kindern der Normalzustand, solange sie gesund sind und regelmäßig Nahrung aufnehmen. Der ganze Körper ist auf Wachstum, Kräftigung und Reifung ausgerichtet.
Jeder kennt aber auch die Zeiten der Krankheiten, in denen genau das Gegenteil der Fall ist. Man liegt nur noch schlapp und müde im Bett, hat keinen Appetit mehr und kann, z.B. während eines Magen-Darm-Infekts, auch so gut wie keine Nahrung mehr bei sich behalten.
Wenn dem Körper somit nicht mehr genügend Nahrung zugeführt wird, muss er notwendigerweise an seine Reserven gehen. Die Muskelmasse und die im Fettgewebe gespeicherten Fette werden nach und nach wieder abgebaut und zu der in den Organen und besonders im Gehirn so dringend benötigten Energie umgewandelt. Der Stoffwechsel funktioniert jetzt gewissermaßen in genau der umgekehrten Richtung. Der ganze Körper schaltet auf Abbau um. Diesen Vorgang nennt man Katabolismus.
In dieser Abbau-Phase kann sich jetzt ein MCAD-Mangel negativ auswirken. Deshalb ist oft davon die Rede, dass es während einer katabolen Stoffwechsellage (wenn sich der Körper also gerade in der Abbauphase befindet) zu einer Krise kommen kann.
Eine katabole Phase kann aber auch ohne Krankheit entstehen, z.B. während einer aufgrund des Schlankheitswahnes selbst auferlegten Hungerdiät, einer ärztlich angeordneten längeren Nüchternphase vor einer Operation, oder einfach einer sehr langen Pause zwischen zwei Mahlzeiten, z.B. zwischen Abendessen und Frühstück.
Die Energiegewinnung während der Abbauphase geschieht zunächst durch die Verwertung der in der Leber und in den Muskelzellen gespeicherten Glykogen-Reserve. Deren Aufgabe besteht normalerweise hauptsächlich darin, den Blutzuckerspiegel zwischen den einzelnen Mahlzeiten auf annähernd konstantem Niveau zu halten. Beim erwachsenen Menschen kann das Glykogen einen Energiegehalt von ca 1900kcal speichern (zum Vergleich: die im Fettgewebe gespeicherte Energiemenge beträgt dagegen etwa 130.000kcal). Betrachtet man die aus Glykogen freisetzbare Energiemenge isoliert, ist sie beim Erwachsenen nach maximal 16 Stunden aufgebraucht. Um diese Reserve nicht zu schnell zu verbrauchen, werden nach einigen Stunden auch die Fettspeicher mobilisiert. Bei der Zerlegung des Fettes in seine Bestandteile entstehen wieder die bereits oben erwähnten Fettsäuren. Diese bestehen aus kettenförmig aneinander gereihten Kohlenstoffeinheiten unterschiedlicher Länge. Kurzkettige Fettsäuren enthalten nur 4-6 Kohlenstoffeinheiten, mittelkettige haben eine Länge von 8-12 und langkettige bestehen aus mehr als 12 Einheiten. Das in den meisten natürlichen Nahrungsmitteln enthaltene Fett besteht ebenso wie das Körperfett hauptsächlich aus langkettigen Fettsäuren mit einer Länge von 18 bis 20 Kohlenstoffeinheiten (oder 16 bis 18, je nachdem, welche Infobroschüre man zu Rate zieht). Auch dabei gibt es ein paar Ausnahmen. Kokosfett und Palmkernfett beispielsweise bestehen zwar auch überwiegend aus langkettigen, jedoch auch zu einem beachtlichen Teil aus mittelkettigen Fettsäuren und ihre flüssigen Varianten Kokosöl und Palmkernöl dienen der Industrie als Rohstoff zur Herstellung von reinem MCT-Fett. Mehr dazu im Artikel zur Ernährung.
Zur Energiegewinnung aus den Fettsäuren werden diese durch verschiedene Enzyme (u.a. die “Dehydrogenasen”) schrittweise um 2er Einheiten verkürzt. Diese 2er Einheiten (Acetyl-CoA) werden in anderen Stoffwechselzyklen weiterverarbeitet, können aber auch Ketonkörper bilden, die direkt als Energielieferanten für das Gehirn dienen.
Die vier Dehydrogenasen
An dem Prozess der Fettsäurenverarbeitung sind neben einer ganzen Reihe anderer Enzyme mit Unterfunktionen auch die vier in Folge arbeitenden “Dehydrogenasen” VLCAD (Very Long Chain Acyl-CoA Dehydrogenase), LCAD (Long Chain Acyl-CoA Dehydrogenase), MCAD (Medium Chain Acyl-CoA Dehydrogenase) und SCAD (Short Chain Acyl-CoA Dehydrogenase) beteiligt. Das VLCAD-Enzym war Anfang der 90er Jahre noch unbekannt, daher wurden damals noch einige Menschen fälschlicherweise mit dem LCAD-Mangel diagnostiziert, und es stellte sich erst Jahre später heraus, dass sie in Wirklichkeit von einem VLCAD-Mangel betroffen waren.
Diese vier Enzyme haben in Bezug auf die Fettsäurenketten unterschiedliche Wirkungsbereiche. Jedoch ist es nicht so, dass es zwischen den einzelnen Enzymen feste Grenzen gibt, auch wenn das in einigen vereinfachten Beschreibungen so dargestellt wird. Jedes Enzym hat seinen Hauptwirkungsbereich mit der größten Aktivität, überlagert aber mit seinen “Ausläufern” auch noch einen Teil der Funktionsbereiche der Vorgänger- und Nachfolger-Enzyme.
Also nicht so:
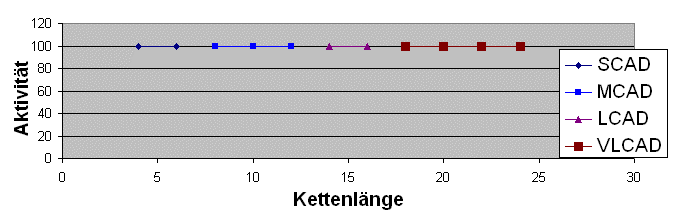
sondern so:
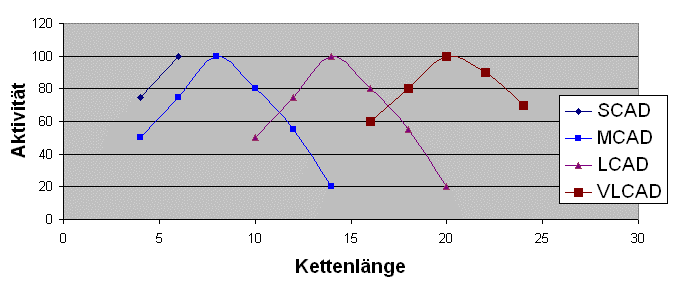
So beginnt die Verarbeitung der Fettsäuren mit VLCAD, welches sich hauptsächlich auf Kettenlängen mit mehr als 16 Kohlenstoffeinheiten konzentriert, jedoch hat es auch im näheren Bereich kleiner gleich 16 noch eine gewisse Restaktivität. Es folgt LCAD mit einem großen Funktionbereich von 10 bis 18 Kohlenstoffeinheiten, danach MCAD mit Aktivität von 4er bis 14er Länge, wobei sich die Hauptaufgabe auf den Bereich von 6 bis 10 konzentriert, und die Verarbeitung endet mit SCAD für 4er und 6er Ketten. Die Grafiken oben sind allerdings nur schematische Darstellungen zur Verdeutlichung der Überlagerungen, und nicht die genau ermittelten Enzymaktivitäten bei den entsprechenden Kettenlängen.
Erblich bedingte Veränderungen in diesen Enzymen führen zum VLCAD-, MCAD- oder SCAD-Mangel.
Das “unbekannte” LCAD-Enzym
Unbekannt deshalb, weil dieses Enzym in der früheren MCAD-Broschüre von Milupa, die den meisten MCAD-Patienten von Seiten der Stoffwechselambulanzen als einziges Informationsmaterial ausgehändigt wurde (und anscheinend auch einigen Stoffwechselärzten als alleinige Informationsgrundlage zum MCAD-Mangel diente), sowie einigen anderen Zusammenfassungen nicht erwähnt wird. Trotzdem existiert es, und nimmt, wie im vorangegangenen Abschnitt beschrieben, seine Funktion in der Aufspaltung von Fettsäuren mit 18er bis 10er Länge wahr. Die Fettsäuren werden daher nicht, wie in der MCAD-Broschüre dargestellt, in drei gleich großen Abschnitten (SCAD, MCAD und VLCAD) mit je 6 Kohlenstoffeinheiten verarbeitet, auch wenn dies für den Einstieg ins Thema eine akzeptable Vereinfachung der Sachverhalte darstellt. Um die Mechanismen und Zusammenhänge jedoch genauer nachvollziehen zu können, muss man die Aufteilung in 4 Dehydrogenasen und deren überlappende Wirkungsbereiche betrachten.
Aufgrund der Tatsache, dass sich mit der Entdeckung des VLCAD-Enzyms in den 90er Jahren die bis dahin als LCAD-Mangel diagnostizierten Fälle als VLCAD-Mangel-Fälle herausstellten, gibt es bisher weltweit keine einzige Person mit eindeutig nachgewiesenem “echtem” LCAD-Mangel. Ebenso wurden − mangels betroffener Patienten − bislang keine spezifischen Mutationen in dem zugrunde liegenden Gen identifiziert. Der Umstand, dass es tatsächlich keinen Menschen mit klinisch auffälligem LCAD-Mangel zu geben scheint, ist für die Mediziner erstaunlich, da es schließlich kritische, oder zumindest zu Auffälligkeiten führende Mutationen in allen anderen Mitgliedern dieser Genfamilie und somit auch entsprechende Stoffwechselstörungen gibt. Dies hat unter Wissenschaftlern die Frage aufgeworfen, welche Rolle das LCAD-Enzym in der Verarbeitung der langkettigen Fettsäuren wirklich spielt, und ob an dieser Stelle eine Stoffwechselstörung überhaupt entstehen kann. Manche Mediziner gehen sogar so weit, aufgrund des Fehlens echter LCAD-Mangel-Fälle die Existenz einer LCAD-Dehydrogenase gänzlich zu bezweifeln. Genau deshalb beschränken sich einige Darstellungen der mitochondrialen Abläufe auf die Aufführung von VLCAD, MCAD und SCAD (siehe frühere MCAD-Broschüre von Milupa), während andere wissenschaftliche Darstellungen (auch neuen Datums!) LCAD in diesen Ablauf sehr wohl einreihen. Die Existenz dieser Dehydrogenase ist seit Jahrzehnten nun mal eindeutig belegt. An Labormäusen − bei denen es einen LCAD-Mangel aufgrund gezielter Züchtung auch wirklich gibt − wurden die unterschiedlichen Auswirkungen von VLCAD- und LCAD-Mangel auch schon untersucht.
Erst unter Berücksichtung dieses zusätzlichen LCAD-Enzyms ist auch verständlich, weshalb anstelle eines C12- oder sogar C14-Wertes erst ein stark erhöhter C8-Wert für die Diagnose des MCAD-Mangels ausschlaggebend ist: die sonst noch in den MCAD-Bereich fallenden C10 bis C14 können zumindest zum Teil auch noch vom LCAD-Enzym aufgespalten werden, und erst auf die 8er Ketten kann nur noch das MCAD-, jedoch nicht mehr das LCAD-, und noch nicht das SCAD-Enzym einwirken.
Die Auswirkung des MCAD-Mangels
Das Problem bei Menschen mit MCAD-Mangel besteht nun darin, dass ihr Körper nicht oder nur sehr eingeschränkt in der Lage ist, das MCAD Enzym zu bilden, oder dass das erzeugte Enzym nur eine sehr eingeschränkte Leistungsfähigkeit aufweist. Es handelt sich um einen angeborenen genetischen Defekt, der fest in der DNA, dem Bauplan des Körpers, programmiert ist.
Somit endet die Zerlegung der Fettsäuren und die damit verbundene Energiegewinnung weitgehend nach dem Wirken von VLCAD und LCAD bei einer Kettenlänge von 8 Einheiten, also etwa der Hälfte des normalen Vorganges. Dies betrifft sowohl den Abbau der Fettsäuren aus dem gespeicherten Körperfett, als auch den Abbau des mit den Mahlzeiten aufgenommenen Nahrungsfettes. In letzterem Fall fällt die auf die Hälfte beschränkte Energiegewinnung aus dem Fett jedoch nicht ins Gewicht, weil üblicherweise gleichzeitig genügend Kohlenhydrate aufgenommen werden, um die Organe mit dem daraus gewonnenen Zucker zu versorgen.
Carnitinmangel − ein gefährlicher Nebeneffekt
Die Aufspaltung der Fettsäuren (ß-Oxidation) innerhalb der Zellen erfolgt in den sogenannten Mitochondrien. Dort befinden sich die dafür benötigten unterschiedlichen Enzyme. Etwa 2000 dieser winzigen Kraftwerke kommen in jeder einzelnen Körperzelle vor. Die Mitochondrien sind von zwei Membranen umschlossen, die jedoch nur von kurz- und mittelkettigen Fettsäuren unmittelbar durchdrungen werden können. Damit (sehr-)langkettige Fettsäuren in die Mitochondrien gelangen können, müssen sie an einen Transporterstoff gekoppelt werden − das Carnitin. Carnitin wird sowohl durch die Nahrung, vor allem beim Verzehr von Fleisch aufgenommen, als auch im Körper selbst gebildet. Nach der Durchdringung der Mitochondrienmembranen wird das Carnitin im Normalfall wieder abgetrennt und steht für weitere Transporte zur Verfügung. Da die Fettsäuren aber infolge des MCAD-Mangels nicht vollständig abgebaut werden können, müssen die übrig gebliebenen Reste der aktivierten Fettsäuren in Form der 12er, 10er und 8er Kohlenstoff-Ketten die Mitochondrien, die Zellen und schließlich auch den Körper unbedingt wieder verlassen. Auch dafür wird wieder Carnitin benötigt, da die verbleibenden mittelkettigen Fettsäuren die Membran der Mitochondrien sonst nicht nach aussen durchdringen könnten. Die Fettsäuren gehen mit dem Carnitin eine chemische Verbindung ein, können dann gemeinsam mit ihm die Zellen verlassen, mit dem Blut zu den Nieren transportiert und dann über den Urin ausgeschieden werden. Bei Menschen mit MCAD-Mangel enthält der Urin daher größere Mengen von an Carnitin gekoppelte mittelkettiger Fettsäurenreste (Acylcarnitin), was bei Menschen ohne MCAD-Mangel nur in sehr geringem Umfang der Fall ist.
Das an die Fettsäuren-Reste gekoppelte Carnitin verlässt den Körper somit vollständig und steht danach nicht mehr innerhalb des Stoffwechsel-Regelkreises zur Verfügung. Wird es nicht in ausreichendem Maße ersetzt, kann sich daraus u.U. ein sekundärer Carnitinmangel entwickeln. Durch den, wie gerade beschrieben, ohnehin erhöhten und besonders in katabolen Phasen besonders stark gesteigerten Carnitinbedarf bei Menschen mit einem MCAD-Mangel, können die im Körper vorhandenen Vorräte schnell zur Neige gehen. In Folge dessen können einerseits nicht mehr genügend langkettige Fettsäuren in die Mitochondrien eingeschleust werden, um wenigstens noch für eine geringe Energiegewinnung zu sorgen, und andererseits werden die Fettsäurenreste nicht mehr aus den Zelle abtransportiert, stauen sich in den Mitochondrien an, blockieren ihre Funktion und führen schließlich zur Entstehung von auf das zentrale Nervensystem toxisch wirkenden Stoffwechselzwischenprodukten, bzw. sogar zum Absterben der Zelle. Während einer Stoffwechselentgleisung geschieht dies in großem Umfang, was die oftmals sehr schweren bleibenden Schädigungen des Gehirns nach einer nicht frühzeitig, bzw. nicht korrekt behandelten Entgleisung erklärt. Bei einem Menschen mit MCAD-Mangel sollen deshalb weitere Untersuchungen von Blut und Urin zeigen, ob zur Vermeidung eines drohenden Carnitin-Mangels auch eine tägliche Ration Carnitin, z.B. in Form eines Sirups, gegeben werden sollte. Über die Nützlichkeit dieser Maßnahme waren und sind sich die Experten seit langer Zeit uneinig. In manchen Stoffwechselambulanzen ist die vorsorgliche unterstützende Gabe einer täglichen kleinen Dosis Carnitin eine der ersten verordneten Maßnahmen, wenn bei einem Neugeborenen ein MCAD-Mangel diagnostiziert wird. Mehr zu diesem Thema im Artikel “Die Behandlung des MCAD-Mangels” ![]()
Ich möchte an dieser Stelle noch einmal ausdrücklich darauf hinweisen, dass ich als Autor dieser Seiten größten Wert auf Korrektheit gelegt habe, aber ansonsten medizinischer Laie bin. Alle veröffentlichten Informationen wurden von mir anhand verschiedener verfügbarer Promotionen, Studienarbeiten, medizinischen Veröffentlichungen, MCAD-Broschüren, Merkblätter, Webseiten von Kliniken, Aussagen der Stoffwechselambulanz und sonstigen Erkenntnissen und Erfahrungen (eigenen und denen unserer Forumsteilnehmer) zusammengetragen und in inzwischen weit über 1000 Stunden Arbeitszeit gelesen, unzählige Male überdacht, sortiert und schließlich in der jetzigen Form hier niedergeschrieben. Das Lesen dieser Seiten darf aber auf keinen Fall den Besuch beim Arzt oder der Stoffwechselambulanz ersetzen, sondern soll lediglich dazu dienen, schon mal etwas besser über die ganze Thematik und Problematik Bescheid zu wissen!